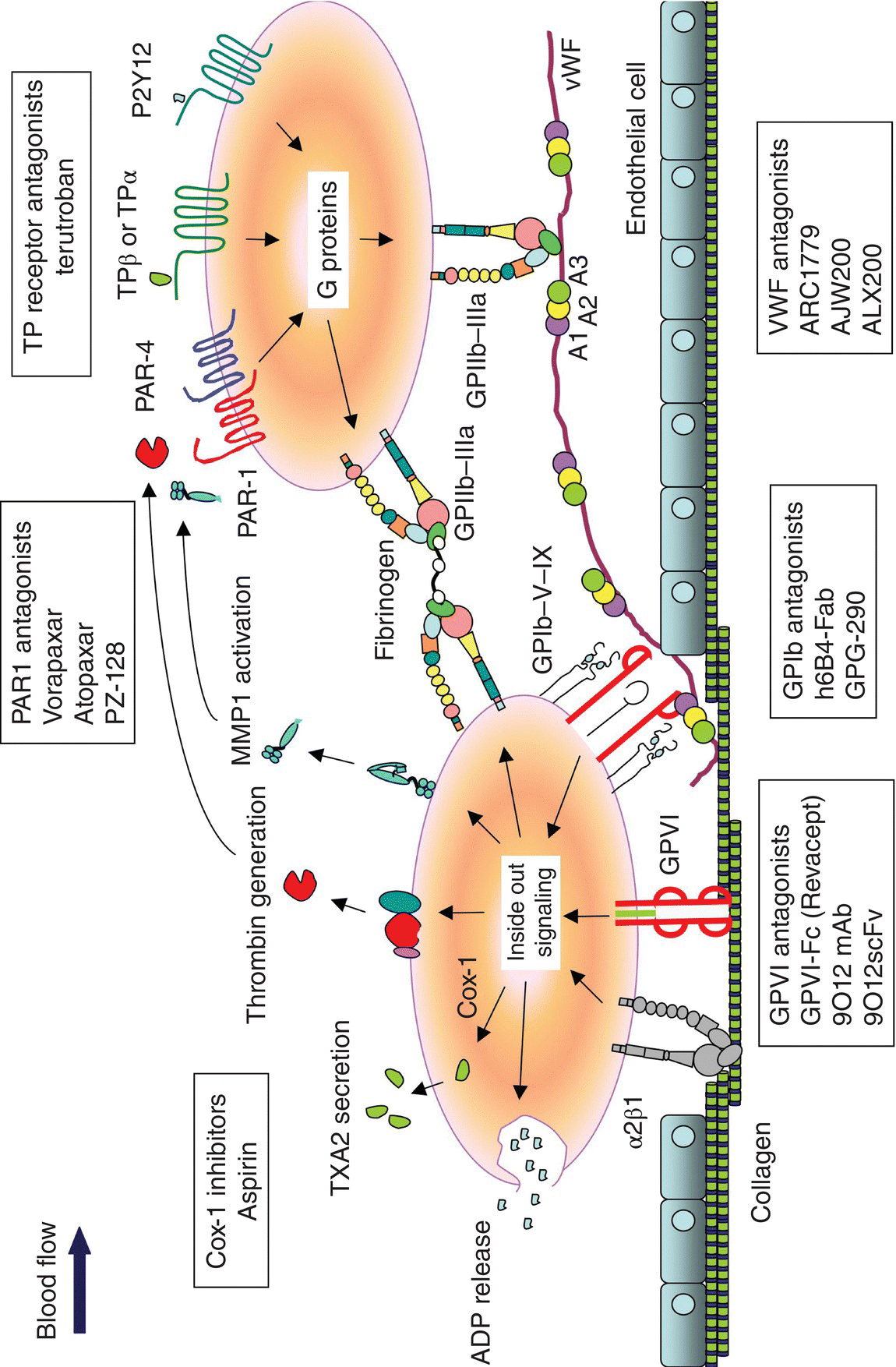
5 Ping Zhang, Lidija Covic, and Athan Kuliopulos Tufts University School of Medicine, Boston, MA, USA Atherothrombotic disease and acute arterial thrombosis are the leading causes of acute coronary syndromes (ACS) and cardiovascular death [1]. Platelets comprise the major component of the arterial thrombi, and effective antiplatelet therapy is critical to prevent ischemic complications of acute arterial thrombosis. Current treatment of patients undergoing percutaneous coronary interventions (PCI) and secondary prevention of ischemic events is a combination of dual antiplatelet therapy consisting of acetylsalicylic acid (ASA, aspirin) to block thromboxane synthesis and a P2Y12 adenosine diphosphate (ADP) receptor inhibitor such as clopidogrel [2]. Dual antiplatelet therapy attenuates short- and long-term ischemic event occurrence during ACS and PCI; however, the persistence of cardiovascular events and the increased risk of bleeding remain major concerns [3, 4, 5, 6, 7, 8]. The most potent P2Y12 receptor blockers are associated with only a 20% relative risk reduction with 10% of patients still suffering from recurrent ischemic events within 1 year of treatment [4, 5]. This indicates that there may be a ceiling effect in a strategy limited to aspirin and P2Y12 receptor inhibition for the prevention of thrombosis in ACS and PCI patients. Intravenous GPIIb–IIIa inhibitors (abciximab, eptifibatide, tirofiban), which block the final common pathway of platelet activation and aggregation, exhibit potent antiplatelet activity but also significantly increase the risk of bleeding proportional to their potency [9]. Therefore, the new platelet inhibitors described in the succeeding text may provide alternative therapeutic efficacy versus hemorrhagic profiles to provide added benefit to patients suffering from life-threatening ischemic arterial disease. The serine protease thrombin is the most potent platelet activator and plays a critical role in thrombosis and in the maintenance of hemostasis following vascular injury [10]. As direct thrombin inhibitors block thrombin-mediated cleavage of fibrinogen, targeting the downstream platelet thrombin receptors instead should theoretically result in a safer bleeding profile. Of the four PAR family members, only PAR1 and PAR4 are expressed on human platelets (Figure 5.1) and can form heterodimers on the platelet surface, which together account for the entire thrombin signal in platelets [11]. Thrombin activates the PARs by cleaving a specific peptide bond in the receptor extracellular domain: R41–S42 for PAR1 and R47–G48 for PAR4 [12]. The signal from the high-affinity PAR1 thrombin receptor is fast and transient [13], whereas the lower-affinity PAR4 [12] evokes a prolonged signal that contributes to irreversible platelet aggregation [11, 13, 14]. PAR1 is the major receptor for thrombin in human platelets and has become an intensively studied antiplatelet target in clinical trials. Conversely, PAR4 is considered to be a secondary thrombin receptor due to its lower affinity for thrombin, and there has been considerably less development of PAR4 inhibitors [11, 13, 15]. More recent studies have identified other proteases such as matrix metalloprotease-1 (MMP1), MMP13, plasmin, and activated protein C (APC) as direct activators of PAR1 [16]. Collagen activation of MMP1–PAR1 signaling may be an important new mechanism of platelet activation during the early steps of platelet thrombogenesis at the site of vessel injury [17]. PAR1 is also important in the vascular remodeling processes in unstable atherosclerotic plaques and in restenosis that may occur after PCI [10, 16, 18]. Chronic blockade of PAR1 in patients with ACS and atherothrombotic disease may therefore provide suppressive effects on both platelets and in culprit lesions undergoing pathological remodeling and inflammation. Figure 5.1 Mechanisms involved in platelet activation and emerging antiplatelet drugs. Following disruption of an atherosclerotic lesion, platelets initially adhere to exposed collagen and von Willebrand factor (vWF) from the vessel wall via GPIb–V–IX on the surface under high shear force conditions. Collagen mediates firm adhesion of platelets in a two-step mechanism in which “outside-in” signaling from the collagen receptor, GPVI, and the α2β1 integrin results in the formation of a platelet monolayer. This collagen-mediated adhesion and activation of platelets leads to the release of adenosine diphosphate (ADP) and thromboxane A2 (TXA2) production via COX-1, matrix metalloprotease-1 (MMP1) activation, and thrombin generation on the surface of activated platelets. These autocrine mediators recruit additional platelets through the major fibrinogen receptor GPIIb–IIIa and activate nearby platelets to cause platelet aggregation via G protein-coupled receptors (GPCRs), PAR1, PAR4, TP, and P2Y12. A number of PAR1 antagonists have been developed including SCH 530348 [19], E5555 [20], FR-17113 [21], F16618 [22, 23], F16357 [24], PZ-128 [25], RWJ-56110 and RWJ-58259 [26, 27, 28], and BMS-200261 [29]. Vorapaxar (SCH 530348) and atopaxar (E5555) have been extensively evaluated in phase III and phase II clinical trials, respectively. Vorapaxar (SCH 530348), an orally active, synthetic analog of himbacine with high affinity (Ki = 3–8 nM) [30] to PAR1, has an exceptionally long elimination half-life of 6.6–13 days [31] and a functionally irreversible binding mode [32]. The pharmacodynamic half-life typically exceeds the entire lifespan of a circulating platelet with 50% recovery of platelet function by 4–8 weeks after a single 20 or 40 mg loading dose of vorapaxar [33]. The onset of inhibition of vorapaxar on PAR1 activity occurs by 2 h after receiving the loading dose. The lowest 10 mg dose gave 43% inhibition of TRAP (PAR1 peptide agonist)-induced platelet aggregation at 2 h, whereas a 40 mg dose resulted in 96% inhibition within 2 h [31]. The safety and tolerability of vorapaxar were evaluated in a phase II randomized trial, Thrombin Receptor Antagonist Percutaneous Coronary Intervention (TRA-PCI), comparing three vorapaxar oral loading doses 10, 20, and 40 mg versus placebo plus standard of care in 1030 patients undergoing nonurgent PCI [34]. The PCI cohort also received unfractionated heparin, low-molecular-weight heparin, or bivalirudin and a loading dose of clopidogrel (300–600 mg) and aspirin (162–325 mg oral or IV 150–500 mg). Patients who underwent PCI were further randomized to one of the three oral daily maintenance doses of vorapaxar (0.5, 1.0, 2.5 mg, or placebo). After 60 days of maintenance of vorapaxar, greater than 80% platelet inhibition to TRAP was seen at all three doses. Patients were evaluated at 60 days for safety (primary end point TIMI major/minor bleeding) and efficacy (MACE – nonfatal MI, nonfatal stroke, hospitalization for recurrent ischemia, or urgent coronary revascularization). The incidence of TIMI major and minor bleeding was similar in all three vorapaxar cohorts as compared to patients receiving placebo, with a dose-dependent trend toward more bleeding at the higher doses. Although not significant, vorapaxar appeared to reduce the occurrence of periprocedural myocardial infarctions (MIs) when added to dual antiplatelet therapy in patients who received PCI [34]. The safety and efficacy of vorapaxar were also evaluated in Japanese patients (n = 117) with a history of non-ST-segment elevation (NSTE) ACS who were receiving standard-of-care therapy (aspirin, ticlopidine, and heparin) [35]. Patients were randomized to receive a loading dose (20 or 40 mg) or placebo and daily oral maintenance doses (1 or 2.5 mg) or placebo for 60 days post-PCI. The primary end point was bleeding (TIMI criteria), and the exploratory end point included all-cause death and MACE (nonfatal MI, nonfatal stroke, hospitalization for recurrent ischemia, or urgent coronary revascularization). Periprocedural MI was defined as an elevated CK-MB or troponin-I (above three times the upper limit of normal with >50% increase above baseline) that was measured at baseline, 8, 16, and 24 h after PCI. Vorapaxar did not result in excess bleeding in the Japanese patients with NSTE ACS and significantly (p = 0.013) reduced the incidence of periprocedural MI by 2.5-fold in subjects undergoing PCI. The majority of MIs that did occur were asymptomatic elevations of CK-MB and troponin-I that were documented during the periprocedural period shortly after PCI. The safety of vorapaxar was evaluated in Japanese patients (n = 90) with a history of ischemic cerebral infarction [36]. All patients received 1.0 or 2.5 mg vorapaxar or placebo once daily for 60 days plus aspirin (75–150 mg/day). The primary end point was overall incidence of adverse events (AE excluding MACE). The AE rate was not significantly different with the dual vorapaxar/aspirin regimen at either dose. The secondary end point of bleeding (TIMI categorized) was similar between placebo and vorapaxar [36]. The PAR1 antagonist atopaxar (E5555) is a second orally active small molecule that inhibits PAR1 activation on platelets. Atopaxar has a much shorter half-life (22–26 h) [37] than vorapaxar. E5555 nearly completely inhibited platelet activation at 20 ng/mL and caused 10–15% inhibition of ADP- and collagen-dependent platelet activation [20]. E5555 also inhibited the expression of the platelet inflammatory markers P-selectin and CD40 ligand. Atopaxar was investigated in several phase II clinical trials including Lesson from Antagonizing the Cellular Effect of Thrombin Acute Coronary Syndromes (LANCELOT) ACS trial (n = 603) [37] and two smaller trials with a Japanese ACS (UA and NSTEMI) patient population (J-LANCELOT) (n = 241) [38] and in patients with coronary artery disease (CAD) (LANCELOT-CAD) (n = 263) on top of standard antiplatelet therapy [39]. ACS patients were pretreated with a 400 mg loading dose of E5555 or placebo followed by 50, 100, or 200 mg daily atopaxar for 12 weeks. Subjects with CAD were treated for 24 weeks without a loading dose with daily 50, 100, and 200 mg atopaxar or placebo on top of aspirin. The LANCELOT-ACS subjects reached maximal platelet inhibition 6 h after the loading dose of atopaxar. The primary end point was the occurrence of bleeding (CURE and TIMI), and the secondary end point was MACE including cardiovascular death, MI, and recurrent ischemia. The LANCELOT-ACS trial results demonstrated that atopaxar significantly reduced Holter-detected ischemia without a clear increase in bleeding compared with placebo. Atopaxar resulted in more minor bleeding in the 200 mg daily dose group and numerically, but not statistically significant, fewer ischemic events in patients with CAD [39]. Atopaxar was generally well tolerated; however, a transient rise in liver enzymes was observed in 3–6% of subjects (p < 0.0001), and prolongation of the QTc interval was observed in some individuals at the higher dose levels [37, 39]. Phase III studies will be required before the efficacy and safety of atopaxar can be fully determined. The efficacy and long-term safety of vorapaxar over 1–2.5-year treatment periods were evaluated in two large phase III randomized trials. The effects of vorapaxar in preventing MI and stroke in patients (n = 26,449) with atherothrombotic disease (either post-MI, a history of stroke, or peripheral arterial disease) were assessed in the Thrombin Receptor Antagonist TRA 2°P-TIMI 50 study [40]. In addition, a second trial assessed the ability of vorapaxar to prevent MI and stroke in patients (n = 12,944) with chronic ACS (TRACER) [41]. Due to elevated bleeding rates, TRA 2°P-TIMI 50 was terminated early in patients that had experienced a stroke before or during the trial (17% of the enrolled patients) [40], and TRACER was terminated in all patients due to significantly increases in the risk of moderate and severe bleeding, including intracranial hemorrhage [42]. When added to standard of care in patients with non-ST elevation ACS and high use of aspirin and P2Y12 inhibition, vorapaxar did not significantly reduce the composite of cardiovascular death, MI, stroke, hospitalization for ischemia, or urgent revascularization. However, vorapaxar did reduce the secondary end point of cardiovascular death, MI, or stroke with a significantly increased incidence of bleeding, including major bleeding and intracranial hemorrhage [42]. For patients with stable atherothrombotic disease (<75 years old and >60 kg), however, inhibition of PAR1 with a loading dose of 40 mg followed by daily 2.5 mg treatment with vorapaxar decreased the risk of cardiovascular death or ischemic events by 13% (p < 0.001) when added to standard dual antiplatelet treatment [40]. The subgroup of patients with prior MI also demonstrated a reduction in cardiovascular death and ischemic events [43]. However, the long half-life of 6–13 days for vorapaxar and only 50% recovery of platelet function at 4 weeks after treatment discontinuation [44] may pose a bleeding risk, especially for patients that may need to undergo subsequent coronary artery bypass (CABG) surgery. Patients with prior MI appear to be most likely to have a net clinical benefit from vorapaxar therapy, and Merck is requesting regulatory approval in patients with prior MI and no history of stroke in the USA and Europe. The ability to rapidly and reversibly inhibit PAR1 signaling by a parenteral strategy would be an ideal in the high-risk patient undergoing PCI. Establishing rapid receptor blockade with a parenterally administered PAR1 blocker should translate into a superior reduction in adverse post-PCI thrombotic event occurrence. Similarly, the use of a more reversible agent than vorapaxar may also facilitate patient care by attenuating bleeding risk in the setting of unanticipated surgery. A fast-acting, shorter half-life pepducin-based drug, PZ-128, is currently being evaluated in human phase I studies. Pepducins are lipidated peptides that target the cytoplasmic surface of their cognate receptor [45, 46]. The PAR1 pepducin, PZ-128 (P1pal-7), is administered by an intravenous infusion during the PCI procedure. PZ-128 consists of a seven-amino-acid peptide derived from the third intracellular loop of PAR1 that is conjugated to palmitate lipid [10, 15, 45]. The solution structure of PZ-128 was determined by NMR, and the peptide was found to form a well-defined α-helix extending from the palmitate lipid. PZ-128 was found to form a highly similar structure as the corresponding region of PAR1 (residues 307–313) in the off-state with an RMSD of 1.4 Å [25]. PZ-128 showed excellent dose- and concentration-dependent inhibition of PAR1 platelet aggregation and arterial thrombosis in baboons. At the 3 mg/kg dose in baboons, PAR1-dependent aggregation was inhibited by 85% at the 1 h and 2 h time points, but was not appreciably inhibited (10%) at the 24 h time point. At the 6 mg/kg dose in baboon, PAR1-dependent aggregation was inhibited by 100% at the 1 h and 2 h time points, 90% at 6 h, but was completely recovered by the 24 h time point. Inhibition of PAR1 by PZ-128 was reversible, as evidenced by loss of inhibition with higher concentrations of SFLLRN agonist at both the 3 mg/kg and 6 mg/kg doses. PZ-128 gave no significant inhibition (0–10%) of either ADP or PAR4 platelet responses at any time point including at 1, 2, or 24 h [25]. Dose-dependent protection against arterial thrombosis with an EC50 of 0.075 mg/kg PZ-128 was determined in guinea pig, and synergistic protective effects were observed with oral clopidogrel. The pharmacokinetic and antiplatelet pharmacodynamic properties of PZ-128 indicate that this lipopeptide reaches maximal activity during (<15 min) and immediately after intravenous infusion and is completely eliminated from plasma by the next day. PZ-128 had no effect on bleeding or coagulation parameters in primates or in blood from PCI patients [25]. The rapid onset of platelet inhibition and reversible properties of PZ-128 are well suited to the acute interventional setting of PCI and may provide an alternative to long-acting small-molecule inhibitors of PAR1 during PCI.
Platelet Receptors and Drug Targets: PAR1, Collagen, vWF, Thromboxane, and Other Novel Targets
Introduction
Protease-activated receptor 1 (PAR1)
Collagen
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


