(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
The ability of thyroid hormone to regulate energy utilization was first recognized more than 100 years ago and is still a contemporary topic in thyroid research. To date, we still do not understand the molecular events by which thyroid hormone controls this essential function. Energy, in the form of ATP, is the currency required for life, and the mitochondrion is its principal source. This fact has made this organelle an obvious target for thyroid hormone action. In addition to providing about 90 % of the energy currency of the cell, these acquired organelles also play essential roles in cell signaling and cell survival, two other cell regulatory cascades that are possible targets for thyroid hormone action.
In addition to the action of the nuclear-localized thyroid hormone receptors on nuclear oxidative phosphorylation (OXPHOS) gene transcription, a parallel direct action of the mitochondrially localized receptors on mitochondrial transcription has been demonstrated. The coordination of transcription activation in nuclei and mitochondria by the thyroid hormone receptors is in part realized by their binding to common trans-acting elements in the two genomes.
In this chapter we will focus on the effects of thyroid hormone on mitochondrial energetics and mitochondriogenesis.
Introduction
Thyroid hormone triiodothyronine (T3) regulates cardiac function through binding to its nuclear receptor (TR). A decrease in T3–TR binding, such as that occurs in systemic hypothyroidism, certain resistance to thyroid hormone syndromes, or in various transgenic or knockout mouse models, adversely affects cardiac function. Furthermore, changes in TR ligand-binding capacity have been reported in failing human hearts.
Classically, the ligand-bound receptors in turn bind to thyroid receptor elements (TRE) and control transcription of key cardiac genes. In the ligand unbound, inactivated state, the TRs together with heat shock and immunophilin chaperones are components of a macromolecular complex. Upon ligand binding, their conformation changes; they are released from the complex in an activated form, dimerize, and interact in the nucleus with respective DNA sequences, TREs. This leads to recruitment of several regulatory proteins, such as coactivators (CoAs) or corepressors (CoRs), some containing intrinsic histone-modifying enzyme activities, and to alteration of chromatin structure, thereby facilitating or blocking the access of the transcriptional machinery to DNA.
Recent data suggest a new paradigm for thyroid hormone regulation, through “nongenomic” or “nonnuclear” T3 actions. This model does not necessarily preclude participation by TRs, as these receptors control key enzyme pathways within the cytosol and mitochondria through protein–protein interactions.
Although abnormalities in TR functioning lead to cardiac dysfunction in part through alterations in excitation–contraction and transport proteins, thyroid also mediates changes in myocardial energy metabolism. Thyroid receptor dysfunction may limit the heart’s ability to shift substrate pathways and provide adequate energy supply during stress responses. Chronic or intermittent energy starvation during these stress periods contributes to abnormal compensatory processes and/or cardiomyocyte damage. Recent data show that T3 also rapidly regulates myocardial metabolism and TR dysfunction alters both substrate flux and signaling by peroxisome proliferator-activated receptors (PPARs). However, the molecular mechanisms of these pathways still require further elucidation.
Thyroid Hormone: Mechanisms of Regulation of Cardiac Mitochondria
Genomic Actions of Thyroid Hormone: Regulation of Expression of Nuclear-Encoded Mitochondrial Proteins
Thyroid hormone through the TR can induce expression of mitochondrial proteins by direct activation of nuclear genes containing thyroid hormone response elements (TREs) in their regulatory sites. They can also modulate expression of mitochondrial proteins indirectly by induction of TRE-containing nuclear genes encoding transcription factors [e.g., nuclear respiratory factors (NRF) 1 and 2].
Direct Regulation of Expression of Nuclear-Encoded Mitochondrial Proteins by Thyroid Hormone Receptor
TRs are members of the nuclear receptor superfamily, which function as T3-inducible transcription factors. They are encoded by two genes located on two different chromosomes. The TRα gene encodes one T3-binding TRα1 and two splicing variants (TRα2 and TRα3), which have no T3-binding activity. TRα1 is constitutively expressed during embryonic development; TRα1 and TRα2 are expressed at lower levels in the heart.
The TRβ gene encodes three T3-binding TRβ isoforms (β1, β2, and β3). TRβ1 is the predominant isoform in the heart, whereas TRβ2 and TRβ3 are minor TRβ isoforms in the myocardium.
The spectrum of TR isoforms is further increased by the addition of receptor molecules produced by alternative translation initiation of the message and by posttranslational modification of the proteins by phosphorylation, acetylation, methylation, ubiquitination, and sumoylation.
Highly conserved DNA-binding domain located centrally in the molecules of thyroid hormone receptor interacts with thyroid hormone response elements on the promoters of T3 target genes. The carboxyl-terminal ligand-binding domain (LBD) binds thyroid hormones, interacts with receptor coregulators (CoRs and CoAs), and also is involved in homodimerization of DNA-bound TRs and heterodimerization with retinoid X receptor (RXR) (Fig. 22.1).
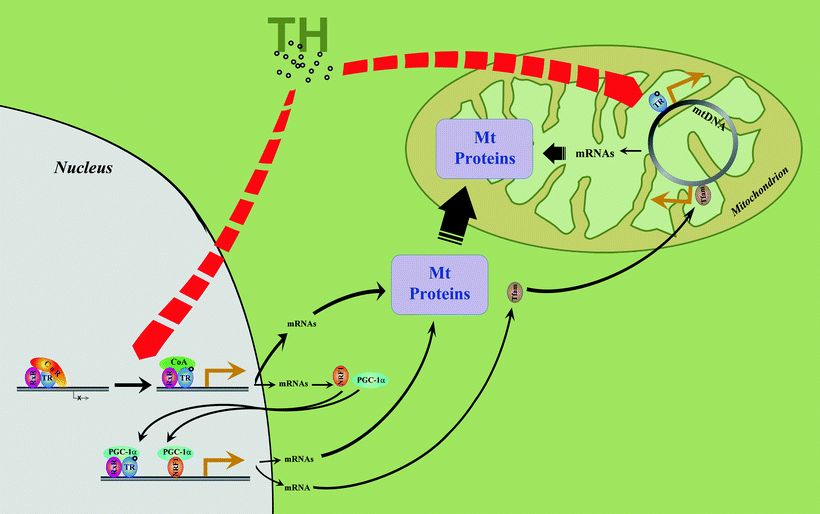
Fig. 22.1
Thyroid hormone-mediated regulation of mitochondrial protein expression. In the nucleus, binding of thyroid hormone (TH) to thyroid hormone receptor (TR) leads to dissociation of receptor corepressor (CoR) and binding of receptor coactivator (CoA). This results in the relief of basal transcriptional repression and induction of transcription. Active TR homodimers or heterodimers with retinoid X receptor (RxR, shown) in the nucleus can directly induce genes of nuclear-encoded mitochondrial proteins (Mt Proteins) having TREs in their regulatory regions. Among TH-regulated genes are nuclear respiratory factors (NRF1 is shown), and peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Their protein products re-enter the nucleus and regulate a second series of TH-regulated mitochondrial target genes. Also TH directly, and via NRF1 and PGC-1α, induces mitochondrial transcription factor A (TFAM). TFAM, a classical genomic TH target gene, regulates mitochondrial DNA (mtDNA) replication and transcription. Yet another level of regulation of mitochondrial protein expression involves binding of TH to mitochondria-located N-terminally truncated TR isoform. Mitochondrial TR specifically binds to TRE-like sequences recorded in the mitochondrial genome and acts as a ligand-dependent mitochondrial transcription factor
Previous studies have shown that several CoRs exist that repress basal TR-dependent transcription of genes in the absence of thyroid hormone: nuclear receptor CoR (NCoR) [1], silencing mediator of retinoid and thyroid hormone receptor (SMRT) [2], Hairless [3], Alien [4], receptor-interacting protein 140 (RIP-140) [5], and small unique nuclear CoR (SUN-CoR) [6]. CoRs associate with other proteins such as transducin-like protein [transducin beta-like protein 1 (TBL1) or TBL1-related protein 1 (TBL1R)] and histone deacetylase 3 (HDAC3) to form large complexes repressing TR via interaction with it [7]. Interestingly, SMRT inhibits PPAR activity and depresses OXPHOS and mitochondrial function. The recent data have also revealed a role for SMRT in aging and the related metabolic disorders [8].
A number of CoAs are also identified that bind to the ligand-bound TR and serving as mediators of activation of TR-dependent gene expression: thyroid hormone receptor-interacting protein 1 (Trip1) [9], steroid hormone receptor CoA 1 (SRC-1) [10], SRC-2 [other names: transcription intermediary factor 2 (TIF2), glucocorticoid receptor-interacting protein 1 (GRIP1)], steroid hormone receptor CoA (SRC) 3 [other names: thyroid hormone receptor activator molecule 1 (TRAM-1), receptor-associated CoA 3 (RAC3), activator of thyroid receptor (ACTR), histone acetyltransferase p300 (p300)/CREB-binding protein (CBP) cointegrator-associated protein (pCIP)] [11], and TR-associated proteins (TRAPs) [12, 13].
Other important findings include:
Mice lacking TRα1 have a lower heart rate, abnormal heart contractility, and decreased body temperature [14].
Gene encoding α-isoform of M-CPTI is regulated by thyroid hormone through elements in the promoter and first intron [15], which leads to changes in both M-CPT1 mRNA abundance and its activity [16]. Interestingly, α- and β-isoforms of M-CPT1 are regulated differently in the rat heart [17]. TRE has been identified in the malic enzyme gene [18].
Another mitochondrial enzyme transcriptionaly regulated by T3 is flavin adenine dinucleotide (FAD)-linked glycerol-3-phosphate dehydrogenase (mGPDH) [19]. It is possible that the rapid induction of mGPDH is based on a direct interaction of the TR with TREs in the mGPDH promoter region.
T3 transcriptionally upregulates pyruvate dehydrogenase (PDH) kinase (PDK) 4, which phosphorylates and inhibits PDH [20, 21].
Thyroid hormone transcriptionally stimulates adenine nucleotide translocator (ANT) in cardiac mitochondria [19, 22, 23], cytochrome c [24], and cytochrome c 1 [25]. The delayed time course of induction of these proteins and lack of TRE sequences in the promoter regions of rat cytochrome c and c 1 [25, 26] raise a question whether T3 directly stimulates transcription via TREs in the promoter region of these genes. It seems more likely that yet unidentified trans-acting factor is involved in the induction of above mentioned genes by thyroid hormone.
TR as a Coregulator of Other Nuclear Receptors that Control Expression of Nuclear-Encoded Mitochondrial Proteins
TRs bind to TREs not only as homodimers but also as heterodimers with other members of the receptor superfamily, such as RXRs, vitamin D receptor, and all subtypes of the retinoic acid receptors.
TR cross talks with PPAR. PPAR regulates the expression of its target genes by binding to the “direct repeat with a 1 bp spacer” PPAR response element (DR1), as a heterodimer with RXR (PPAR–RXR). Heterodimer TRβ–RXR competes with PPAR–RXR to repress the transcriptional activity of PPARγ [27]. For instance, transgenic mice, expressing the dominant negative TRβ1 specifically in the heart, show altered cardiac transcriptional response to PPARα agonist [28]. Because PPAR plays a key role in lipid metabolism and cardiovascular diseases [29, 30], this mode of TR action via cross talk with other receptors expands TR effects via direct binding to TREs on the promoter of target genes.
TR-Dependent Regulation of Expression of Other Nuclear Receptors that Control Expression of Nuclear-Encoded Mitochondrial Proteins
Thyroid hormone regulates NRF1 [31], an essential nuclear regulator of several OXPHOS genes such as β- and γ-subunits of ATP synthase, and complex IV subunits Vb, VIc, and VIIa [32], and mitochondrial transcription factor A (TFAM), which in turn regulates the rate of transcription of the mitochondrial genome (see below).
Thyroid Hormone Regulates Expression of Mitochondria-Encoded Proteins
Thyroid hormone regulates the expression of the structural genes of mitochondrial DNA (mtDNA), such as cytochrome c oxidase (COX) subunit 1, cytochrome b, and several mtDNA-encoded subunits of NADH dehydrogenase (ND 1, 4, and 5). T3-dependent regulation appears to be at the transcriptional level (increase of mRNAs) [36]. There are several molecular mechanisms of mitochondrial gene’s transcriptional activation by T3, including increases in mitochondrial RNA polymerase or in nuclear factors, which regulate promoter recognition by the polymerase or direct regulation of mitochondrial genes.
TR-Dependent Regulation of Nucleus Genes Encoding Mitochondrial Transcription Factors
Thyroid hormone regulates two nuclear-encoded general mitochondrial transcription factors, PGC-1 [31, 33] and TFAM [37–39]. The gene TFAM is located on human chromosome 10q21. TFAM is imported into mitochondria where it regulates the rate of transcription of the mitochondrial genome via promoters in a D-loop region of mtDNA responsible for initiation of global increases in mitochondrial gene expression. Evidences for essential role of TFAM for mtDNA replication and transcription have been obtained by transfection of monkey kidney cells with an expression vector containing TFAM in antisense orientation: transection leads to reduced expression of TFAM and inhibited transcription of mtDNA [40]. Similarly, heterozygous TFAM knockout in mice reduced mtDNA copy number and lessened cardiac mitochondrial electron transport chain (ETC) capacity [41]. Mice with conditional knockout of cardiac TFAM show cardiac-specific OXPHOS deficiency, conduction block, dilated cardiomyopathy, and die at 2–4 weeks of age [42]. The promoter region of TFAM does not appear to contain TREs, but expression of TFAM is regulated by T3 indirectly via NRF (Fig. 22.1).
T3 increases mitochondrial import of the nuclear-encoded mitochondrial heat shock protein (mtHsp)-70, and the increased matrix level of this chaperone enhances import of other mitochondrial proteins from the cytosol [43].
Thyroid Hormone: Direct Regulation of Transcription of Mitochondrial Genes
Recently truncated forms of the TRα [44–46] and TRβ1 [47] were shown to import to the mitochondrial matrix because they are encoded by transcripts originating from an internal AUG start sites in TR genes and lack N-terminal nuclear targeting A/B domains.
Once inside the mitochondria, truncated TRs interact with two authentic TREs located in the D-loop, an element of the mitochondrial genome, to initiate global increases in mitochondrial gene expression. TREs are also located in the mitochondrial genes encoding the 12S and 16S rRNA in mitochondria [44]. Thus, the N-terminal truncated TRs serve as T3-dependent transcription factors that initiate global mitochondrial transcription. Full-length TRs have also been identified within cardiac mitochondria [46–48].
Experimental proof for a direct action of thyroid hormones on mitochondrial transcription has been provided by Enriquez et al. [49] and of Casas et al. [45], using an in organello mitochondrial system from rat liver. Hypothyroid rat liver mitochondria have shown a decrease in the mRNA/rRNA ratio compared to that of euthyroid controls. In vitro addition of thyroid hormone to mitochondria from hypothyroid animals has restored the mRNA/rRNA ratio [49]. Targeting the mitochondrial-specific truncated TR (p43) to mitochondria leads to increased levels of precursor and mature RNA and of the mRNA/rRNA ratio in a thyroid hormone-dependent manner, when in organello mtDNA transcription system has been used [45].
Thus, the mitochondrial TR is a mitochondrial, thyroid hormone-activated transcription factor, which would exert its modulatory effect by interaction with the mitochondrial transcriptosome (Fig. 22.1).
Thyroid Hormone as a Nongenomic Regulator of Mitochondrial Protein/Enzyme Activities
ANT has been implicated as a direct receptor for T3 in cardiac mitochondria [50–52]. Initially, thyroid hormone binding component of mitochondrial inner membrane (MIM) was described by Sterling and Milch in 1975 [53] and later confirmed in several other laboratories [54, 55]. Physicochemical characteristics of putative mitochondrial T3 receptor (a mass and amino acid composition) were similar to mitochondrial ANT [56, 57], which is known to be regulated by thyroid hormone [58, 59]. ANT purified from beef heart mitochondria has been found to exhibit high-affinity, low-capacity binding of T3 [50]. Moreover, ANT inserted into artificial proteoliposomes demonstrates displaceable high-affinity binding of T3. These findings make the mitochondrial ANT a strong candidate for nongenomic regulatory site for thyroid hormone stimulation in mammalian species.
Xenopus xTRβA1, which is similar to mitochondrial targeted truncated TRα, increases mitochondrial membrane potential, possibly through interactions with components of electron transport chain. Also xTRβA1 inhibits cytochrome c release in T3-dependent fashion leading to a fall in apoptotic activity in vitro and in CV1 cells and enhances inositol-1,4,5-trisphosphate (IP3)-mediated calcium signaling [60, 61].
An unusual mechanism involving indirect action of thyroxine (T4) has been suggested for regulation of mitochondrial monoamine oxidase (MAO). This enzyme is bound to the mitochondrial outer membrane (MOM) and is involved in the inactivation of neurotransmitters via oxidation of monoamines. It is well documented that the thyroid hormone affects metabolism of catecholamines by altering activity of MAO [62–64]. MAO activity is markedly inhibited in cardiac mitochondria 1 day later after administration of T4 to rats and stays inhibited for another 4 days [65]. These decreases of MAO activity are not due to changes of MAO protein levels and do not depend on the direct inhibition by T4. Interestingly, heart cytosolic fraction from T4-treated rats inhibits mitochondrial MAO activity, suggesting T4-dependent expression of cytosolic protein modulator(s) that change MAO catalytic activity [65, 66].
Thyroid Hormone and Bioenergetics: Regulation of Mitochondrial Energy Production
Mitochondrial respiration is the most efficient way of producing energy in the form of adenosine triphosphate (ATP). In mitochondria, reducing equivalents produced by the tricarboxylic acid (TCA) cycle or coming from the β-oxidation of fatty acids (FAs) are oxidized by the multienzyme respiratory chain (complexes I–IV). Oxidation consumes oxygen to produce water, whereas protons are pumped out of mitochondrial matrix to create a proton gradient. The electrochemical gradient across the MIM drives ATP synthase to produce ATP. The key sources of reducing equivalents utilized in the heart are free FAs (FFAs), glucose, and lactate. They represent the majority of oxidative fuel directed into the mitochondria to enter TCA cycle.
Cellular energy demands dictate adaptive changes in energy production by the mitochondria. Depending on the challenge, mitochondria can change oxidative capacity modulating the activity of OXPHOS or biosynthesis of some of the OXPHOS components. In addition, the number and size of the organelles also can significantly be changed (see following section). Therefore, mitochondria, as major sources of ATP generation, are the targets for thyroid hormone-mediated regulation of cardiac metabolism [67, 68]. Goldenthal et al. [69] have demonstrated that major components of cardiac mitochondrial bioenergy-producing machinery are regulated by T3.
It is well documented that cytochrome c oxidase enzyme is upregulated in response to T3 treatment [70, 71]. In mammals, COX is composed of 13 polypeptides, ten of which are transcribed within the nucleus, and the remaining three subunits are products of the mitochondrial genome. In the myocardium, T3 induces mitochondria-encoded COX1 expression, and it parallels the T3-induced increases in COX activity. [69, 70] Levels and activity of COX1 (and also nuclear-encoded COX4) are significantly elevated shortly after in vitro treatment of neonatal rat cardiomyocytes with triiodothyronine (3–12 h) [69]. Surprisingly, while nuclear gene encoding another subunit of cytochrome c oxidase, COX5b, contains TRE in promotor region, it is not regulated by T3 in the heart [70], suggesting a role for TR corepressors, limiting the induction of this gene in the heart. Instead of influencing the expression of COX5b, a unique nongenomic mechanism of interaction of thyroid hormones with this subunit has been described. In particular, diiodothyronines (and T3 to lesser extent) have been shown to bind specifically to COX5b protein from bovine heart [72, 73]. This binding abolishes the allosteric inhibition of cytochrome c oxidase activity by ATP. Direct interaction of thyroid hormones with COX5b explains at the molecular level the short-term stimulatory action of thyroid hormones on mitochondrial respiration (Fig. 22.2). Yet another mechanism of regulation of cytochrome c oxidase by thyroid hormones acts through mitochondrial phospholipid, cardiolipin. According to Paradies et al. [74], cytochrome c oxidase activity is decreased in heart mitochondria from hypothyroid rats, and activity of this enzyme complex can be completely restored to the level of control rats by exogenously added cardiolipin.
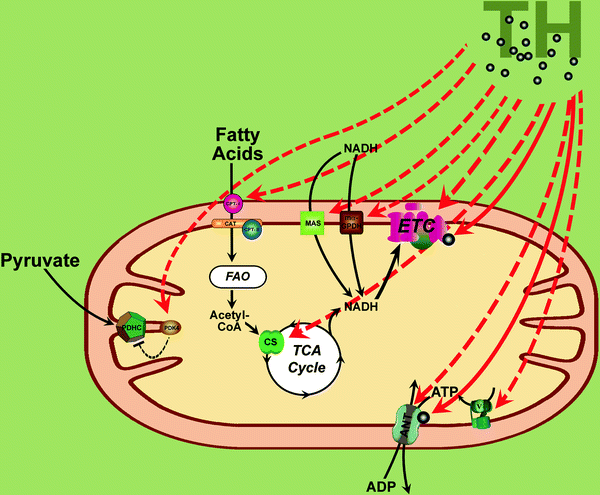
Fig. 22.2
Thyroid hormone and mitochondrial energetics. Thyroid hormones (TH) regulate multiple genes involved in energy metabolism extending from those contributing to substrate oxidation, the respiratory chain, and oxidative phosphorylation to genes regulating mitochondrial ATP and NADH transport and glucose and fatty acid oxidation. TH-dependent induction of electron transport chain (ETC) components (complexes I through IV, cytochrome c) and ATP synthase (V) via coordinated expression of nuclear- and mitochondria-encoded genes is shown by dashed red arrows. Similarly, transcriptional regulation by TH of fatty acid transporting system, pyruvate dehydrogenase complex (PDHC), citrate synthase (CS), adenine nucleotide translocator (ANT), and NADH-transporting shuttles (MAS malate/aspartate shuttle, mα-GPDH mitochondrial α-glycerophosphate dehydrogenase) is shown by dashed red arrows. In mitochondria TH may bind/activate subunit 5b of cytochrome c oxidase and ANT which is thought to directly increase oxidative phosphorylation and cause the rapid thermogenic response to T3 (shown by solid red arrows). See text for further details. Other abbreviations: Acetyl-CoA acetyl coenzyme A, CAT carnitine acyltransferase, CPTI carnitine palmitoyltransferase I, CPTII carnitine palmitoyltransferase II, FAO fatty acid oxidation, PDK4 PDH kinase 4, TCA Cycle tricarboxylic acid cycle
Cytochrome c oxidase is not the only ETC component sensitive to thyroid hormones. For instance, gene encoding subunit 3 (ND3) of NADH dehydrogenase (also known as complex I) has been found to contain TRE. Transcriptional regulation of this gene by thyroid hormones in the heart has been demonstrated by Iglesias et al. [75]: level of ND3 mRNA is significantly diminished in hypothyroid rats compared to control animals. Remarkably, gene encoding ND3 is located in mtDNA, suggesting direct transcriptional regulation of ND3 by thyroid hormone via TRs identified inside the mitochondria (see previous section of this chapter). In vitro treatment of neonatal rat cardiomyocytes with triiodothyronine also elevates level and activity of ATP synthase (complex V) α-subunit (early response, 3–12 h) and activities of complex II and citrate synthase (late response, at 72 h) [69]. Apparently, long-term T3 effect on mitochondria is complex, involving regulation of other mitochondrial proteins in addition to ETC. For example, it is accompanied by induction of ANT [69] and uncoupling protein (UCP) 3 [76], which cause an uncoupling in mitochondria [19, 77] with subsequent less efficient energy production in the cardiomyocyte (Fig. 22.2).
In addition to respiratory-chain components, proteins that connect cytosolic and mitochondrial metabolic pathways play a key role in the T3-mediated modulation of energy metabolism. Thus, the expression of ANT type 2 mRNA is increased seven- to ninefold in rat heart within 12–48 h after T3 application. These mRNA changes correlate with an increase of ANT protein, which explains the accelerated ADP/ATP exchange observed in mitochondria isolated from hyperthyroid rats [19]. Two myocardial transport pathways mediate transport of cytosolic reducing equivalents (in form of NADH) to the mitochondrial OXPHOS system. The malate/aspartate shuttle is the dominant NADH-transporting pathway in cardiomyocytes, whereas α-glycerophosphate shuttle is tenfold less active. The NADH shuttles have a potential to optimize bioenergy substrate utilization in hyperthyroid-induced cardiac hypertrophy when increased glycolytic flux takes place instead of utilization of FAs. The malate/aspartate shuttle capacity is significantly increased in ventricular mitochondria isolated from T3-treated rats. In particular, protein levels of two components of the malate/aspartate shuttle system, mitochondrial malate dehydrogenase (mMDH) and aspartate/glutamate carrier (AGC), are found to be increased in the thyroid hormone-treated myocardium, suggesting that either protein could contribute to the increased capacity of the malate/aspartate shuttle. Taking into account that state of mMDH is the near equilibrium, the increased levels of AGC carrier seem to be responsible for the upregulation of the malate/aspartate shuttle in the thyroid hormone-treated cardiac mitochondria [78]. While regulation of mMDH gene by thyroid hormone is posttranscriptional (changes in mMDH protein levels are not accompanied by changes in mRNA), it is possible that the regulation of AGC gene expression is directly regulated by T3. Transport of cytosolic NADH to the mitochondria via the glycerophosphate shuttle is very sensitive to T3: mRNA of the FAD-linked mitochondrial α-glycerophosphate dehydrogenase (mα-GPDH), key component of this shuttle, is induced very fast (4–6 h) by T3 in rat heart [19, 78, 79], and mα-GPDH protein levels increase in parallel [78]. These findings suggest that thyroid hormone directly stimulates nuclear transcription of the mα-GPDH gene (Fig. 22.2).
Key enzyme regulating glucose oxidation in mitochondria—pyruvate dehydrogenase complex (PDHC)—is also a target for thyroid hormone. Chronic T3 supplementation in rats decreases activity of PDH. It has been shown that T3 transcriptionally upregulates PDK4, which phosphorylates and inhibits PDHC [20, 21] (Fig. 22.2). It should be mentioned that a controversy exists between effects of T3 on pyruvate oxidation in mitochondria and on pyruvate uptake by mitochondria: while oxidation of pyruvate is inhibited (via T3-dependent regulation of PDK4/PDH), transport of this molecule into mitochondria increases [80]. Transport of pyruvate across mitochondrial membrane is mediated by mitochondrial pyruvate carrier, and Paradies and Ruggiero [80] have demonstrated that triiodothyronine significantly enhances this process. It is possible that in hyperthyroid cardiac mitochondria, increased pyruvate converts into oxaloacetate by mitochondrial pyruvate carboxylase (rather than oxidized via inhibited PDH) to support the oxidation (via TCA cycle) of acetyl coenzyme A (acetyl-CoA) derived from fatty acid oxidation (FAO). Interestingly, T3-dependent enhancement of pyruvate uptake does not depend on the increase of pyruvate carrier expression but rather involves positive modulation of the pyruvate carrier activity by cardiolipin [80] (cardiolipin levels in mitochondria are known to increase in hyperthyroid rats—see below).
Another potential target for T3-dependent modulation of substrate oxidation is direct regulation of lactate transport and oxidation in the mitochondria. Lactate generally supplies approximately 25% of myocardial oxidative substrate at normal physiological levels [81, 82]. It enters cardiomyocyte through the monocarboxylate transporter 1 (MCT1) [83] and is oxidized by lactate dehydrogenase within the mitochondria. Recent findings demonstrate colocalization of the MCT1, the MCT1-chaperone (CD147), and lactate dehydrogenase within mitochondrial reticulum of muscle cells [84]. T3 promotes lactate exchange at the cell sarcolemma not via modulation of MCT1 expression but rather via changing its activity [85] and altering translocation of MCT1 and CD147 to the mitochondria.
The major cardiac functional changes induced by hypothyroidism include a drop in heart rate and a decrease in diastolic function, and these alterations are linked to energy metabolism in mitochondria affected by hypothyroidism. In particular, thyroid hormone alters FFA flux into TCA cycle via regulation of mitochondrial FAO. In fact, cardiac mitochondrial FAO is significantly depressed in hypothyroid rats [86], so the contribution of acetyl-CoA entering the TCA cycle via PDHC (lactate, glucose) increases in the hypothyroid state relative to that from FFAs and ketones [67]. The oxidation of long-chain FAs depends on their transport across the MIM mediated by the carnitine palmitoyltransferase (CPT)/carnitine–acylcarnitine translocase system. Depressed respiratory rates correlate with low FA translocation activity in cardiac mitochondria from hypothyroid rats, suggesting T3-related changes in functional activity of FA-transporting system. In agreement with this hypothesis, it has been shown that systemic hypothyroidism in rats decreases expression of muscle-type CPTI (M-CPTI), a central component of the CPT/carnitine–acylcarnitine translocase system, associated with the MOM [67, 68]. In addition, Paradies et al. [86] have demonstrated 38% diminishing of carnitine–acylcarnitine exchange across the MIM of cardiac mitochondria from hypothyroid rats—a process catalyzed by another component of FA-transporting system, carnitine–acylcarnitine translocase (Fig. 22.2).
In the latter case, it has been suggested that the reduced mitochondrial translocase activity in hypothyroid rats could be due to the changes in cardiolipin levels in the lipid surrounding of the carnitine carrier molecule in the MIM. Cardiolipin locates almost exclusively in the MIM where it is biosynthesized [87–90] and has been reported to be specifically required for full functioning of the carnitine–acylcarnitine translocase (see also Chap. 3) [91, 92]. Importantly, cardiolipin content is markedly decreased in the cardiac MIM from hypothyroid rats and can be restored to the level of control rats by treatment of hypothyroid rats with T3 [86]. Interestingly, it has been reported that the biosynthesis of cardiolipin in mitochondria is regulated by thyroid hormone via an alteration in the cardiolipin synthase activity [93, 94]. Thus, decrease of cardiolipin synthase activity and levels of cardiolipin may represent the molecular mechanism responsible for the decline in the activity of mitochondrial FA-transporting system under hypothyroid state.
Thyroid hormone influences uncoupling of substrate oxidation to ATP generation, which produces heat. An uncoupling is achieved by disturbing the proton gradient across the MIM by “proton leak” altering proton trafficking across the MIM or by covalent modification of the ADP/ATP translocator [95].
In rodents, UCP1, which belongs to the superfamily of mitochondrial anion carriers, mediates inducible proton leaks, and catecholamines and thyroid hormone regulate an expression of this protein. It is worth to note that this protein is unique to brown fat tissue, whereas other tissues express UCP2 and UCP3 that do not appear to be regulated by thyroid hormone [96–98]. Outside of brown fat, thyroid hormone regulates basal mitochondrial proton conductance by unknown mechanism [99].
Thyroid hormone plays an important role in postnatal metabolic adaptation of the myocardium. After birth, rise of the thyroid hormone prompts elevations in mRNA and protein level of ANT in cardiac mitochondria (Fig. 22.2). As a result, ADP/ATP exchange across the mitochondrial membrane increases, and cardiac mitochondrial respiration becomes ADP independent: increases in cardiac work are not accompanied by increases in ADP [22, 23].
Thyroid Hormone and Mitochondrial Biogenesis
The heart, which maintains a high oxidative capacity relative to other organs, must respond rapidly to changing energy requirements coming across physiologically or during stress conditions. One way to increase cardiac oxidative capacity and stimulate substrate oxidation pathways on demand is to elevate mitochondrial copy number (or higher mitochondrial mass) because the mitochondrion is a key regulator of the metabolic activity of the cell and is an important organelle in both production and degradation of free radicals. The process, by which new mitochondria are formed in the cell, is called mitochondrial biogenesis.
The expression of genes both in the nuclear and mitochondrial genome is required for mitochondrial production. The majority of mitochondrial protein is synthesized under control of the nuclear genome, while the mitochondrial genome encodes several components of ETC along with mitochondrial rRNA and tRNA. A major adaptive response of mitochondrial biogenesis results in more mitochondrial mass associated with increased glycolytic enzymes, oxidative phosphorylation, and ultimately with a greater mitochondrial metabolic capacity.
The master regulators of mitochondrial biogenesis appear to be the PGC-1 family of transcriptional coactivators, including PGC-1α, PGC-1β, and the PGC-related coactivator, PRC. PGC-1α, in particular, is thought to be a master regulator. It is known to coactivate NRF1 and NRF2 (also known as GABPA). The NRFs, in turn, activate TFAM, which is directly responsible for transcribing nuclear-encoded mitochondrial proteins. This includes both structural mitochondrial proteins as well as those involved in mtDNA transcription, translation, and repair.
The ability of thyroid hormone to increase the number of mitochondria in a cell is well known. In rats chronically treated with thyroid hormone, myocardial mitochondrial mass, mitochondrial respiration, and OXPHOS enzyme activities, mitochondrial protein synthesis and mtDNA levels increase, indicating overall increase in mitochondrial biogenesis [100]. Recent data show that T3 coordinates cardiac mitochondrial and nuclear transcription required for the propagation of mitochondria [69, 100]. Thyroid hormone participates in the synthesis of nuclear-encoded mitochondrial proteins contributing to substrate oxidation, the ETC, and oxidative phosphorylation (the vast majority of mitochondrial proteins) [101]. Studies using microarray techniques have identified approximately 4–8% of randomly selected genes to be regulated by T3 in mammalian tissues [102–106], whereas only a limited number of genes have been proven to be directly regulated by TRs via TREs. Based on the fact that many T3 target genes lack TREs in the regulatory elements together with the data of differential gene expression patterns after T3 treatment (there are “early expression” and “late expression” T3-inducible genes) [103, 104, 107], it appears multiple pathways are involved in T3-mediated gene regulation. For instance, TRE-containing gene of mitochondrial glycerol-3-phosphate dehydrogenase is induced within the first hour after T3 administration in hypothyroid rats in vivo [108]. In contrast, genes that have been induced late (>12 h) after treatment of hypothyroid rats with T3 (ANT2, cytochromes c and c 1, complex II, α- and β-subunits of mitochondrial ATP synthase, citrate synthase, TFAM) [19, 24, 25, 39, 69, 109] are regulated via indirect mechanisms.
Many nuclear genes encoding mitochondrial proteins that respond to T3 in the process of mitochondrial biogenesis contain common DNA motifs in their promoters, which are recognition sites for nuclear regulatory factors, so they serve as intermediate mediators of T3 action. For example, late T3-inducible genes encoding cytochrome c and TFAM have functional sites for binding NRF1 [110–112]. NRF1 plays an essential function in mitochondrial biogenesis [113] and is endogenously regulated by T3, probably via a TRE [104].
Transcription factors Specificity Protein 1 (Sp1) and Ying Yang 1 (YY1) have been demonstrated to be critical factors for their promoter activities of mitochondrial protein genes. Their target genes (ANT2, cytochromes c and c1, COX4, β-subunit of mitochondrial ATP synthase, TFAM) [110–112, 114–116] are regulated by T3 in a late inducible manner, supporting the idea of Sp1 and YY1 as intermediate factors. However, it is currently unknown if Sp1 and YY1 are themselves regulated by T3. Moreover, the DNA-binding proteins targeting the OXBOX/REBOX and Mt motifs in T3-sensitive nuclear-encoded mitochondrial proteins (ANT, β-subunit of mitochondrial ATP synthase) are currently unknown [117–119].
Action of thyroid hormone on the expression of proteins during mitochondrial biogenesis can also be mediated via regulation of coactivators and corepressors of transcription factors—proteins that do not bind directly to DNA but interact with transcription factors and modulate their activity. For instance, PGC-1α is a potent coactivator of nuclear receptors (including TR) and NRF1 [33, 120] and has a profound influence on mitochondrial biogenesis and metabolic rate [33, 121]. T3 promotes PGC-1 protein in cultured neonatal rat myocytes [69]. Importantly, these findings are in agreement with in vivo data: PGC-1α expression levels in hypothyroid rats increase 13-fold 6 h after administration of T3 and remain high for at least 42 h [104]. Two other members of PGC-1 family, PRC and PGC-1β, show some similar binding properties to nuclear receptors [122–124], but it is currently unknown if they are regulated by T3.
Also, level of global regulatory transcription factor PPARα increases in the myocardium of 15-day T4-treated rats as well as in rat cardiomyocytes treated with T3 in vitro [69, 100]. PPARα is implicated in the expression of enzymes catalyzing mitochondrial FAO [125] and an expression of UCP [126]. Thus, the parallel increase in mitochondrial biogenesis and PPARα level suggests the participation of PPARα in the mitochondrial biogenesis as a response to T3.
It is important to highlight that by modulating PGC-1α [33] and TFAM [37–39], T3 serves as a bigenomic coordinator, because these regulatory proteins are involved in mitochondrial transcription. Moreover, various thyroid hormone receptors have been demonstrated to import into mitochondria for binding and transactivation of the mitochondrial genome. In liver, truncated forms of the TRα1 [44, 45] and TRβ1 [47] specifically translocate into the mitochondrion, bind T3, and stimulate generalized transcription of the mitochondrial genome via activation of genes in D-loop of mtDNA and expression of mitochondrial 12S and 16S rRNAs [44]. Full-length TRα1 as well as multiple truncate versions have been identified within cardiac mitochondria [45, 127, 128]. To conclude, the complex finely tuned communication network coordinates the expression of mitochondrial and nuclear genomes to produce new, fully functional mitochondria.
Thyroid hormone appears to target two mitochondrial enzymes involved in the biosynthesis of cardiolipin, one of the principle phospholipids in the mammalian heart. Cardiolipin is the major mitochondrial membrane lipid playing a vital role in mitochondrial function. Phosphatidylglycerolphosphate synthase catalyzes formation of phosphatidylglycerolphosphate which after dephosphorylation serves as a substrate of cardiolipin synthase to synthesize cardiolipin. Administration of thyroxine has been shown to increase phosphatidylglycerol and cardiolipin content in rat ventricular mitochondrial fractions compared to controls (34% and 23% to 72%, respectively) [94, 129, 130]. The activities of phosphatidylglycerolphosphate synthase and cardiolipin synthase parallel T3-induced changes in phospholipids, by increasing 3.5- and 1.5- to 2.8-fold, respectively [93, 94]. Thus, thyroid hormone-dependent stimulation of mitochondrial biogenesis includes increase in mitochondrial phospholipids in addition to mitochondrial proteins, RNA, and DNA. It is worth to note that T3-dependent changes in cardiolipin concentration may underlie alterations in activities of several mitochondrial cardiolipin-sensitive protein systems, such as long-chain FA-transporting system [91, 92], ANT [131], cytochrome c oxidase [74, 132], and pyruvate and phosphate translocators [80, 92, 133].
Process of mitochondrial biogenesis, in addition to coordinated expression of mitochondrial proteins encoded by both the nuclear and mitochondrial genomes, synthesis of lipids, and insertion of nascent proteins in the lipid bilayer, also involves mitochondrial protein import pathway. Mitochondrial proteins that are encoded by nuclear genes are synthesized as precursor proteins on cytosolic polysomes. The precursors harbor mitochondria-targeting sequences, which allow cytosolic chaperones to escort them to mitochondria (see also Chap. 4). Once at the mitochondrion, the precursor interacts with import receptors, collectively called translocases. Translocases are located in both the outer and inner mitochondrial membranes (TOM and TIM, respectively). They guide the precursor in the mitochondrion, where the targeting signal is proteolytically cleaved to form a mature mitochondrial protein [134, 135]. The protein import in mitochondria is influenced by T3 [136], and limited information exists regarding regulation of individual import machinery components by thyroid hormones. Thus, it has been shown that the 20-kDa translocase of the MOM (Tom20) and the matrix chaperone mtHsp70 are upregulated in response to T3 [43, 137] with parallel increase in import of various mitochondrial enzymes [137]. Recently, altered thyroid status has been demonstrated to affect Tom34 levels in the myocardium; T3-dependent changes in the mRNA expression of various components of the Tim machinery in vivo have also been observed [136].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


