John M. Miller, Douglas P. Zipes
Therapy for Cardiac Arrhythmias
Treatment of patients with tachyarrhythmias has evolved dramatically over the last 40 years. Antiarrhythmic drugs were the mainstay of therapy until the late 1960s, when surgical therapy to cure, not just suppress tachyarrhythmias was developed. This mode was replaced by catheter ablation for better control or even cure of tachyarrhythmias in the 1980s. Catheter ablation has largely replaced surgical and drug therapy for patients who need treatment of supraventricular tachycardia (SVT) and ventricular tachycardia (VT) in the absence of structural heart disease. The implantable cardioverter-defibrillator (ICD) was introduced in the early 1980s and has become standard therapy for patients with serious ventricular arrhythmias in the presence of structural heart disease. Some patients require a combination of these forms of treatment (hybrid therapy, such as an ICD and antiarrhythmic drugs or surgery and an ICD); drug therapy can also affect ICD function, either positively or negatively. Drug therapy for arrhythmias, at one time the only option, has largely been replaced as the mainstay of therapy by ablation or implanted devices. However, in most patients, tachyarrhythmias are initially treated with antiarrhythmic drugs, and thus they continue to have a significant role.
Pharmacologic Therapy
The principles of clinical pharmacokinetics and pharmacodynamics are discussed in Chapter 9.
General Considerations Regarding Antiarrhythmic Drugs
Most of the antiarrhythmic drugs available (Table 35-1) can be classified according to whether they exert blocking actions predominantly on sodium, potassium, or calcium channels and whether they block receptors. The commonly used classification, that of Vaughan Williams, is limited because it is based on the electrophysiologic effects exerted by an arbitrary concentration of the drug, generally on a laboratory preparation of normal cardiac tissue. In reality, the actions of these drugs are complex and depend on tissue type, degree of acute or chronic damage, heart rate, membrane potential, ionic composition of the extracellular milieu, autonomic influence, genetics (see Chapter 32), age (see Chapter 76), and other factors (see Table 35-1). Many drugs exert more than one type of electrophysiologic effect or operate indirectly, such as by altering hemodynamics, myocardial metabolism, or autonomic neural transmission. Some drugs have active metabolites that exert effects different from those of the parent compound. Not all drugs in the same class have identical effects (e.g., amiodarone, sotalol, and ibutilide). Whereas all class III agents are dramatically different, some drugs in different classes have overlapping actions (e.g., class IA and class IC drugs). Thus, in vitro studies on healthy myocardium usually establish the properties of antiarrhythmic agents rather than their actual antiarrhythmic properties in vivo.
TABLE 35-1
Actions of Drugs Used for the Treatment of Arrhythmias
| Channels | Receptors | Pumps | Predominant Clinical Effects | |||||||||||
| DRUG | Na* | Ca | Kr | Ks | α | β | M2 | P | Na,K-ATPase | LV FUNCTION | SINUS RATE | EXTRACARDIAC | ||
| Fast | Medium | Slow | ||||||||||||
| Quinidine | ●A | ◉ | ○ | ○ | — | ↑ | ◉ | |||||||
| Procainamide | ●I | ◉ | ↓ | — | ◉ | |||||||||
| Disopyramide | ●A | ◉ | ○ | ↓ | Var | ● | ||||||||
| Ajmaline | ●A | — | — ↓ | ○ | ||||||||||
| Lidocaine | ○ | — | — ↓ | ○ | ||||||||||
| Mexiletine | ○ | — | — | ○ | ||||||||||
| Phenytoin | ○ | — | — | ◉ | ||||||||||
| Flecainide | ●A | ○ | ↓ | — | ○ | |||||||||
| Propafenone | ●A | ○ | ◉ | ↓ | ↓ | ○ | ||||||||
| Propranolol | ○ | ● | ↓ | ↓ | ○ | |||||||||
| Nadolol | ● | ↓ | ↓ | ○ | ||||||||||
| Amiodarone | ○ | ◉ | ● | ◉ | ◉ | ◉ | — | ↓ | ● | |||||
| Dronedarone | ○ | ◉ | ● | ◉ | ◉ | ◉ | — | ↓ | ○ | |||||
| Sotalol | ● | ● | ↓ | ↓ | ○ | |||||||||
| Ibutilide | Activator | ○ | — | ↓ | ○ | |||||||||
| Dofetilide | ● | — | — | ○ | ||||||||||
| Verapamil | ○ | ● | ◉ | ↓ | ↓ | ○ | ||||||||
| Diltiazem | ◉ | ↓ | ↓ | ○ | ||||||||||
| Adenosine | □ | — | ↓ | ◉ | ||||||||||
| Digoxin | ○ | ● | ↑ | ↓ | ◉ | |||||||||
| Atropine | ● | — | ↑ | ◉ | ||||||||||
| Ranolazine | ○ | ○ | — | — | ○ | |||||||||
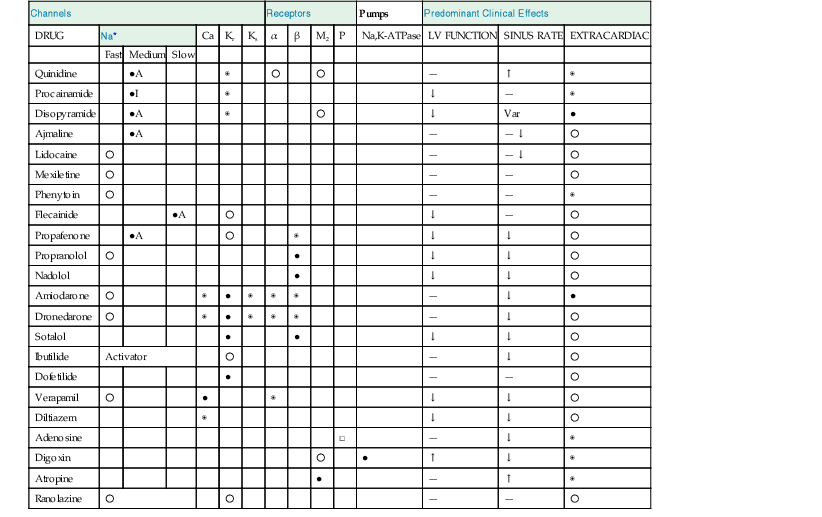
* Fast, medium, and slow refer to the kinetics of recovery from sodium channel blockade.
Relative potency of blockade or extracardiac side effect: ○ = low; ◉ = moderate; ● = high.
□ = agonist; A = activated state blocker; I = inactivated state blocker.
— = minimal effect; ↑ = increase; ↓ = decrease; Var = variable effects.
Kr = rapid component of the delayed rectifier K+ current; Ks = slow component of the delayed rectifier K+ current; M2 = muscarinic receptor subtype 2; P = A1 purinergic receptor.
ATPase = adenosine triphosphatase; LV = left ventricular.
Modified from Schwartz PJ, Zaza A: Haemodynamic effects of a new multifactoral antihypertensive drug. Eur Heart J 13:26, 1992. Copyright © 1992. Reproduced by permission of the publisher W.B. Saunders Company Limited.
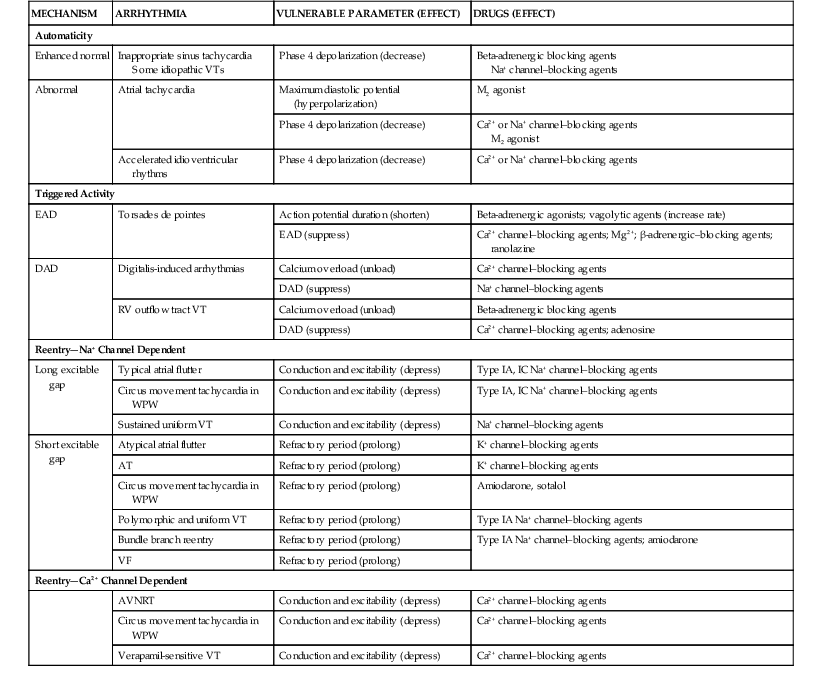
Despite its limitations, the Vaughan Williams classification is widely known and provides a useful communication shorthand, but the reader is cautioned that drug actions are more complex than those depicted by the classification. A more realistic view of antiarrhythmic agents is provided by the Sicilian gambit. This approach to drug classification is an attempt to identify the mechanisms of a particular arrhythmia, to determine the vulnerable parameter of the arrhythmia most susceptible to modification, to define the target most likely to affect the vulnerable parameter, and then to select a drug that will modify the target.1 This concept provides a framework in which to consider antiarrhythmic drugs (Table 35-2; also see Table 35-1).
Drug Classification
According to the Vaughan Williams classification, class I drugs predominantly block the fast sodium channel; they can also block potassium channels. They, in turn, are divided into three subgroups, classes IA, IB, and IC (Table 35-3).
TABLE 35-3
In Vitro Electrophysiologic Characteristics of Antiarrhythmic Drugs
| DRUG | APD | dV/dt | MDP | ERP | CV | PF PHASE 4 | SN Auto | Contr | SI Curr | AUTONOMIC NERVOUS SYSTEM |
| Quinidine | ↑ | ↓ | 0 | ↑ | ↓ | ↓ | 0 | 0 | 0 | Antivagal; alpha blocker |
| Procainamide | ↑ | ↓ | 0 | ↑ | ↓ | ↓ | 0 | 0 | 0 | Slight antivagal |
| Disopyramide | ↑ | ↓ | 0 | ↑ | ↓ | ↓ | ↓ 0 ↑ | ↓ | 0 | Central: antivagal, antisympathetic |
| Ajmaline | ↑ | ↓ | 0 | ↑ | ↓ | ↓ | ↓ 0 | ↓ | 0 | Antivagal |
| Lidocaine | ↓ | 0 ↓ | 0 | ↓ | 0 ↓ | ↓ | 0 | 0 | 0 | 0 |
| Mexiletine | ↓ | 0 ↓ | 0 | ↓ | ↓ | ↓ | 0 | ↓ | 0 | 0 |
| Phenytoin | ↓ | ↓ 0 ↑ | 0 | ↓ | 0 | ↓ | 0 | 0 | 0 | |
| Flecainide | 0 ↑ | ↓ | 0 | ↑ | ↓↓ | ↓ | 0 | ↓ | 0 | 0 |
| Propafenone | 0 ↑ | ↓ | 0 | ↑ | ↓↓ | ↓ | 0 | ↓ | 0 ↓ | Antisympathetic |
| Propranolol | 0 ↓ | 0 ↓ | 0 | ↓ | 0 | ↓* | ↓ | ↓ | 0 ↓ | Antisympathetic |
| Amiodarone | ↑ | 0 ↓ | 0 | ↑ | ↓ | ↓ | ↓ | 0 ↑ | 0 | Antisympathetic |
| Dronedarone | ↑ | 0 ↓ | 0 | ↑ | ↓ | ↓ | ↓ | 0 ↓ | 0 | Antisympathetic |
| Sotalol | ↑ | 0 ↓ | 0 | ↑ | 0 | 0 ↓ | ↓ | ↓ | 0 ↓ | Antisympathetic |
| Ibutilide | ↑ | 0 | 0 | ↑ | 0 | 0 | ↓ | 0 | 0 | 0 |
| Dofetilide | ↑ | 0 | 0 | ↑ | 0 | 0 | 0 | 0 | 0 | 0 |
| Verapamil | ↓ | 0 | 0 | 0 | 0 | ↓* | ↓ | ↓ | ↓↓ | ? Block alpha receptors; enhance vagal |
| Adenosine | ↑ | 0 ↓ | More (−) | ↑ | 0 | 0 ↓ | ↓ | 0 | ↓ | Vagomimetic |
| Ranolazine | ↑ | 0 | 0 | ↑ | 0 | 0 | 0 | 0 | 0 | 0 |
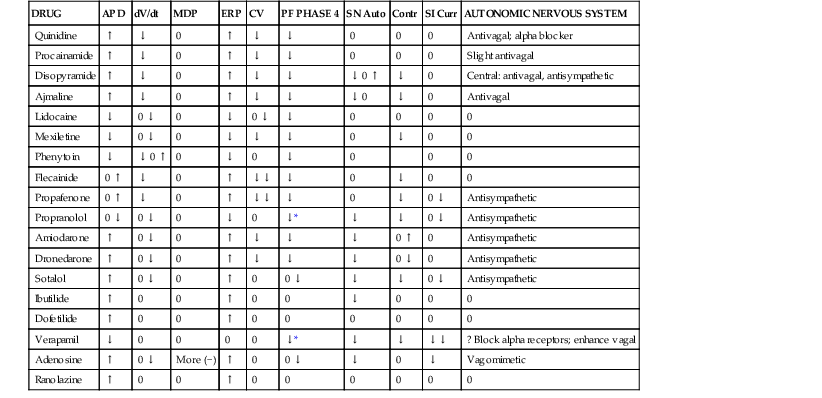
* With a background of sympathetic activity.
dV/dt = rate of rise of action potential; MDP = maximum diastolic potential; ERP = effective refractory period (longest S1-S2 interval at which S2 fails to produce a response); CV = conduction velocity; PF = Purkinje fiber; SN Auto = sinus nodal automaticity; Contr = contractility; SI Curr = slow inward current.
Class IA
This class includes drugs that reduce  max (rate of rise in action potential upstroke [phase 0]) and prolong the action potential duration (APD; see Chapter 33)—quinidine, procainamide, and disopyramide. The kinetics of onset and offset of class IA drugs in blocking the Na+ channel is of intermediate rapidity (less than 5 seconds) when compared with class IB and class IC agents.
max (rate of rise in action potential upstroke [phase 0]) and prolong the action potential duration (APD; see Chapter 33)—quinidine, procainamide, and disopyramide. The kinetics of onset and offset of class IA drugs in blocking the Na+ channel is of intermediate rapidity (less than 5 seconds) when compared with class IB and class IC agents.
Class IB
This class of drugs does not reduce  max and shortens the APD—mexiletine, phenytoin, and lidocaine. The kinetics of onset and offset of these drugs in blocking the sodium channel is rapid (less than 500 milliseconds).
max and shortens the APD—mexiletine, phenytoin, and lidocaine. The kinetics of onset and offset of these drugs in blocking the sodium channel is rapid (less than 500 milliseconds).
Class IC
This class of drugs, including flecainide and propafenone, can reduce  max, primarily slow conduction velocity, and prolong refractoriness minimally. These drugs have slow onset and offset kinetics (10 to 20 seconds).
max, primarily slow conduction velocity, and prolong refractoriness minimally. These drugs have slow onset and offset kinetics (10 to 20 seconds).
Class II
These drugs block beta-adrenergic receptors and include propranolol, metoprolol, nadolol, carvedilol, nebivolol, and timolol.
Class III
This class of drugs predominantly blocks potassium channels (such as IKr) and prolongs repolarization. Included are sotalol, amiodarone, dronedarone, and ibutilide.
Class IV
This class of drugs predominantly blocks the slow calcium channel (ICa.L)—verapamil, diltiazem, nifedipine, and others (felodipine blocks ICa.T).
Antiarrhythmic drugs appear to cross the cell membrane and interact with receptors in the membrane channels when the channels are in the rested, activated, or inactivated state (see Table 35-1 and Chapter 33), and each of these interactions is characterized by different association and dissociation rate constants of a drug on its receptor. Such interactions depend on voltage and time. Transitions among rested, activated, and inactivated states are governed by standard Hodgkin-Huxley–type equations. When the drug is bound (associated) to a receptor site at or very close to the ionic channel (the drug may not actually plug the channel), the channel cannot conduct, even in the activated state.
Use Dependence.
Some drugs exert greater inhibitory effects on the upstroke of the action potential at more rapid rates of stimulation and after longer periods of stimulation, a characteristic called use dependence. Use dependence means that depression of  max is greater after the channel has been “used” (i.e., after action potential depolarization rather than after a rest period). Agents in class IB exhibit fast kinetics of onset and offset or use-dependent block of the fast channel; that is, they bind and dissociate quickly from receptors. Class IC drugs have slow kinetics, and class IA drugs are intermediate. With increased time spent in diastole (slower rate), a greater proportion of receptors become drug free, and the drug exerts less effect. Unhealthy cells with reduced (i.e., abnormal) membrane potentials recover more slowly from drug actions than do healthier cells with more negative (i.e., normal) membrane potentials.
max is greater after the channel has been “used” (i.e., after action potential depolarization rather than after a rest period). Agents in class IB exhibit fast kinetics of onset and offset or use-dependent block of the fast channel; that is, they bind and dissociate quickly from receptors. Class IC drugs have slow kinetics, and class IA drugs are intermediate. With increased time spent in diastole (slower rate), a greater proportion of receptors become drug free, and the drug exerts less effect. Unhealthy cells with reduced (i.e., abnormal) membrane potentials recover more slowly from drug actions than do healthier cells with more negative (i.e., normal) membrane potentials.
Reverse Use Dependence.
Some drugs exert greater effects at slow rates than at fast rates, a property known as reverse use dependence. This is particularly true for drugs that lengthen repolarization. The QT interval becomes more prolonged at slow rather than at fast rates. This effect is not what the ideal antiarrhythmic agent would do because prolongation of refractoriness should be increased at fast rates to interrupt or to prevent a tachycardia and should be minimal at slow rates to avoid precipitation of torsades de pointes.
Mechanisms of Arrhythmia Suppression.
Given the fact that enhanced automaticity, triggered activity, or reentry can cause cardiac arrhythmias (see Chapter 33), mechanisms by which antiarrhythmic agents suppress arrhythmias can be postulated (see Table 35-2). Antiarrhythmic agents can slow the spontaneous discharge frequency of an automatic pacemaker by depressing the slope of diastolic depolarization, shifting the threshold voltage toward zero, or hyperpolarizing the resting membrane potential. Mechanisms whereby different drugs suppress normal or abnormal automaticity may not be the same. In general, however, most antiarrhythmic agents in therapeutic doses depress the automatic firing rate of spontaneously discharging ectopic sites while minimally affecting the discharge rate of the normal sinus node. Slow channel blockers such as verapamil, beta blockers such as propranolol, and some antiarrhythmic agents such as amiodarone also depress spontaneous discharge of the sinus node, whereas drugs that exert vagolytic effects, such as disopyramide and quinidine, can increase the sinus discharge rate. Drugs can also suppress early or delayed afterdepolarizations and eliminate triggered arrhythmias related to these mechanisms.
Reentry depends critically on the interrelationships between refractoriness and conduction velocity, the presence of unidirectional block in one of the pathways, and other factors that influence refractoriness and conduction, such as excitability (see Chapter 33). An antiarrhythmic agent can stop ongoing reentry that is already present or prevent it from starting if the drug depresses or, alternately, improves conduction. For example, improving conduction can (1) eliminate a unidirectional block so that reentry cannot begin or (2) facilitate conduction in the reentrant loop so that the returning wave front reenters too quickly, encounters cells that are still refractory, and is extinguished. A drug that depresses conduction can transform a unidirectional block into a bidirectional block and thus terminate reentry or prevent it from starting by creating an area of complete block in the reentrant pathway. Conversely, a drug that slows conduction without producing block or lengthening refractoriness significantly can promote reentry. Finally, most antiarrhythmic agents share the ability to prolong refractoriness relative to their effects on APD; that is, the ratio of the effective refractory period (ERP) to APD exceeds 1.0. If a drug prolongs the refractoriness of fibers in the reentrant pathway, the pathway may not recover excitability in time to be depolarized by the reentering impulse, and reentrant propagation ceases. The different types of reentry (see Chapter 33) influence the effects and effectiveness of a drug.
In considering the properties of a drug, it is important that the situation or model from which conclusions are drawn be defined with care. Electrophysiologic, hemodynamic, autonomic, pharmacokinetic, and adverse effects may all differ in normal subjects as compared with patients, in normal tissue as compared with abnormal tissue, in cardiac muscle as compared with specialized conduction fibers, and in atrium as opposed to ventricular muscle (Table 35-4).
TABLE 35-4
Clinical Use Information for Antiarrhythmic Agents
| Usual Dosage Ranges | ||||||||||
| DRUG | INTRAVENOUS (mg) | ORAL (mg) | TIME TO PEAK PLASMA CONCENTRATION (ORAL) (hr) | EFFECTIVE SERUM OR PLASMA CONCENTRATION (µg/mL) | HALF-LIFE (hr) | BIOAVAILABILITY (%) | MAJOR ROUTE OF ELIMINATION | PREGNANCY CLASS | ||
| Loading | Maintenance | Loading | Maintenance | |||||||
| Quinidine | 6-10 mg/kg at 0.3-0.5 mg/kg/min | — | 800-1000 | 300-600 q6h | 1.5-3.0 | 3-6 | 5-9 | 60-80 | Liver | C |
| Procainamide | 6-13 mg/kg at 0.2-0.5 mg/kg/min | 2-6 mg/min | 500-1000 | 250-1000 q4-6h | 1 | 4-10 | 3-5 | 70-85 | Kidney | C |
| Disopyramide | 1-2 mg/kg over 15-45 min* | 1 mg/kg/hr* | N/A | 100-300 q6-8h | 1-2 | 2-5 | 8-9 | 80-90 | Kidney | C |
| Lidocaine | 1-3 mg/kg at 20-50 mg/min | 1-4 mg/min | N/A | N/A | N/A | 1-5 | 1-2 | N/A | Liver | B |
| Mexiletine | 500 mg* | 0.5-1.0 g/24 hr* | 400-600 | 150-300 q8-12h | 2-4 | 0.75-2 | 10-17 | 90 | Liver | C |
| Phenytoin | 100 mg q5min for ≤1000 mg | N/A | 1000 | 100-400 q12-24h | 8-12 | 10-20 | 18-36 | 50-70 | Liver | D |
| Flecainide | 2 mg/kg* | 100-200 q12h* | 50-200 q12h | 3-4 | 0.2-1.0 | 20 | 95 | Liver | C | |
| Propafenone | 1-2 mg/kg* | N/A | 600-900 | 150-300 q8-12h | 1-3 | 0.2-3.0 | 5-8 | 25-75 | Liver | C |
| Propranolol | 0.25-0.5 mg q5min to ≤0.20 mg/kg | N/A | N/A | 10-200 q6-8h | 4 | 1-2.5 | 3-6 | 35-65 | Liver | C |
| Amiodarone | 15 mg/min for 10 min 1 mg/min for 3 hr 0.5 mg/min thereafter | 0.5 mg/min | 800-1600 qd for 7-14 days | 200-600 qd | Variable | 0.5-1.5 | 56 days | 25 | Kidney | D |
| Dronedarone | N/A | N/A | N/A | 400 mg q12h | 3-4 | 0.3-0.6 | 13-19 | 70-90 | Liver | X |
| Sotalol | 10 mg over 1-2 min* | N/A | N/A | 80-320 q12h | 2.5-4 | 2.5 | 12 | 90-100 | Kidney | B |
| Ibutilide | 1 mg over 10 min | N/A | N/A | N/A | N/A | N/A | 6 | Kidney | C | |
| Dofetilide | 2-5 µg/kg infusion* | N/A | N/A | 0.125-0.5 q12h | 7-13 | 90 | Kidney | C | ||
| Verapamil | 5-10 mg over 1-2 min | 0.005 mg/kg/min | N/A | 80-120 q6-8h | 1-2 | 0.10- 0.15 | 3-8 | 10-35 | Liver | C |
| Adenosine | 6-18 mg (rapidly) | N/A | N/A | N/A | N/A | N/A | Seconds | 100 | Blood cells | C |
| Digoxin | 0.5-1.0 mg | 0.125-0.25 qd | 0.5-1.0 | 0.125-0.25 qd | 2-6 | 0.0008-0.002 | 36-48 | 60-80 | Kidney | C |
| Ranolazine | N/A | N/A | N/A | 500-1000 bid | 4-6 | N/A | 7 | 60-75 | Liver, Kidney | C |
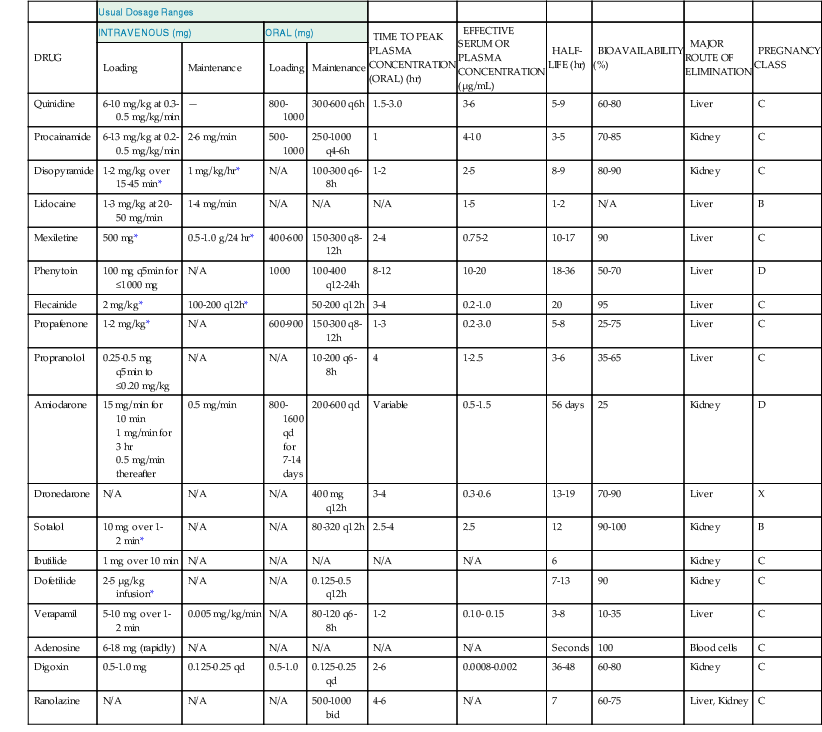
* Intravenous use investigational or unavailable in the United States.
Results presented may vary according to doses, disease state, and IV or oral administration.
Pregnancy class: A = controlled studies show no fetal risk; B = no controlled studies, but no evidence of fetal risk; fetal harm unlikely; C = fetal risk cannot be excluded; drug should be used only if potential benefits outweigh potential risk; D = definite fetal risk; drug should be avoided unless in a life-threatening situation or safer alternatives do not exist; X = contraindicated in pregnancy
N/A = not applicable.
Drug Metabolites.
Drug metabolites can add to or alter the effects of the parent compound by exerting similar actions, competing with the parent compound, or mediating drug toxicity. Quinidine has at least four active metabolites but none with a potency exceeding that of the parent drug and none implicated in causing torsades de pointes. About 50% of procainamide is metabolized to N-acetylprocainamide (NAPA), which prolongs repolarization and is a less effective antiarrhythmic drug but competes with procainamide for renotubular secretory sites and can increase the parent drug’s elimination half-life. Lidocaine’s metabolite can compete with lidocaine for sodium channels and partially reverse block produced by lidocaine.
Pharmacogenetics.
Genetically determined metabolic pathways account for many of the differences in patients’ responses to some drugs (see Chapter 9).2 The genetically determined activity of hepatic N-acetyltransferase regulates the development of antinuclear antibodies and lupus syndrome in response to procainamide. Slow acetylator phenotypes appear to be more prone than rapid acetylators to the development of lupus. The enzyme cytochrome P-450 (CYP450) is needed to metabolize propafenone, to hydroxylate several beta blockers, and to biotransform flecainide. Lack of this enzyme (in ≈7% of patients) reduces metabolism of the parent compound and thereby leads to increased plasma concentrations of the parent drug and reduced concentrations of metabolites. Propafenone is metabolized by CYP450 to a compound with slightly less antiarrhythmic and beta-adrenergic blocking effects, as well as fewer central nervous system side effects. Thus, poor metabolizers may experience more heart rate slowing and neurotoxicity than extensive metabolizers do.
Drugs such as rifampin, phenobarbital, and phenytoin induce the synthesis of larger amounts of CYP450, which leads to lower concentrations of the parent drugs because of extensive metabolism, whereas erythromycin, clarithromycin, fluoxetine, and grapefruit juice inhibit enzyme activity, which leads to accumulation of the parent compound. Cisapride, a gastric motility agent, blocks the delayed rectifier current IKr but does not prolong the QT interval significantly in most patients because of extensive metabolism. In patients who take an inhibitor of CYP450 (such as erythromycin) along with cisapride, the latter drug could accumulate and lead to QT prolongation and torsades de pointes.
Clinical Use
In treating cardiac rhythm disorders, most drugs are given on a daily basis (in one to three doses) to prevent episodes from occurring or, in some cases of atrial fibrillation, to control the ventricular rate. Efficacy can be judged in various ways, depending on the clinical circumstances. Symptom reduction (in the case of benign arrhythmias, such as most premature ventricular complexes [PVCs]) and electrocardiographic monitoring (long-term or event; see Chapter 34) are useful; electrophysiologic studies (EPSs) have been used in the past, with suppression of induction of electrical arrhythmia being the goal. However, this is rarely used currently. Interrogation of implanted device memory can also provide an indicator of the success of drug therapy.
In some patients, tachycardia episodes are infrequent enough (months between occurrences) and symptoms mild enough that reactive drug administration is more reasonable than chronic daily dosing. In this setting a patient takes a medication only after an episode has started in the hope that the tachycardia will terminate in response to the drug and a visit to a physician’s office or emergency department can be avoided. This “pill in the pocket” strategy has worked well for some patients with atrial fibrillation who have been given one of various medications orally in a monitored setting to ensure safety, as well as efficacy, before allowing self-medication at home or elsewhere.
Side Effects
Antiarrhythmic drugs produce one group of side effects related to excessive dosage and plasma concentrations that result in both noncardiac (e.g., neurologic defects) and cardiac (e.g., heart failure, some arrhythmias) toxicity and another group of side effects unrelated to plasma concentrations, which is termed idiosyncratic. Examples of the latter include amiodarone-induced pulmonary fibrosis and some arrhythmias, such as quinidine-induced torsades de pointes, which can occur in individuals with a forme fruste of long-QT syndrome (i.e., normal QT interval at rest but markedly prolonged interval in the presence of certain medications; see Chapters 9 and 32). In the future, it is likely that genetic differences will explain many idiosyncratic reactions.
Proarrhythmia
Drug-induced or drug-exacerbated cardiac arrhythmias (proarrhythmia) constitute a major clinical problem. Proarrhythmia can be manifested as an increase in frequency of a preexisting arrhythmia, sustaining of a previously nonsustained arrhythmia (even making it incessant), or development of arrhythmias that the patient has not previously experienced. Electrophysiologic mechanisms are probably related to prolongation of repolarization or an increase in its transmural dispersion, development of early afterdepolarizations with resultant torsades de pointes, and alterations in reentry pathways to initiate or to sustain tachyarrhythmias (see Chapter 33). Proarrhythmic events can occur in as many as 5% to 10% of patients receiving antiarrhythmic agents. Heart failure increases this risk. Reduced left ventricular function, treatment with digitalis and diuretics, and a longer pretreatment QT interval characterize patients who experience drug-induced ventricular fibrillation (VF). The more commonly known proarrhythmic events occur within several days of beginning drug therapy or changing dosage and are represented by such developments as incessant VT, long-QT syndrome, and torsades de pointes. However, in CAST (Cardiac Arrhythmia Suppression Trial), researchers found that encainide and flecainide reduced spontaneous ventricular arrhythmias but were associated with a total mortality of 7.7%, as opposed to 3.0% in the group receiving placebo. Deaths were equally distributed throughout the treatment period, thus raising the important consideration that another type of proarrhythmic response can occur some time after the beginning of drug therapy. Such late proarrhythmic effects may be related to drug-induced exacerbation of the regional myocardial conduction delay caused by ischemia and to heterogeneous drug concentrations that can promote reentry. In coming years, a candidate antiarrhythmic compound’s potential for proarrhythmia may be modeled computationally or tested in stem cells.3
The availability of catheter ablation (see later) and implantable devices (pacemakers and ICDs; see Chapter 36) to treat a wide variety of arrhythmias has largely relegated drug therapy to a secondary role in the treatment of serious arrhythmias. Drugs are still useful to prevent or to decrease the frequency of recurrences in patients who have relatively infrequent episodes of benign tachycardias, those who have had incomplete success with catheter ablation procedures, and patients with an ICD to decrease the frequency of shocks because of supraventricular or ventricular arrhythmias.
Antiarrhythmic Agents
Class IA Agents
Quinidine
Quinidine and quinine are isomeric alkaloids isolated from cinchona bark.4 Although quinidine shares the antimalarial, antipyretic, and vagolytic actions of quinine, only quinidine has electrophysiologic effects. It blocks several channels (rapid inward sodium channel, IKr, Ito, and to a lesser extent, the slow inward calcium channel, IKs, and the adenosine triphosphate (ATP)-sensitive potassium current [KATP]).
Electrophysiologic Actions.
Quinidine exerts little effect on automaticity of the normal sinus node but suppresses automaticity in normal Purkinje fibers (Table 35-5; see also Tables 35-1, 35-2, and 35-3). In patients with sick sinus syndrome, quinidine can depress sinus node automaticity. Quinidine produces early afterdepolarizations in experimental preparations and in humans, which may be responsible for torsades de pointes. Because of its significant anticholinergic effect and the reflex sympathetic stimulation resulting from alpha-adrenergic blockade, which causes peripheral vasodilation, quinidine can reflexly increase the sinus node discharge rate and improve atrioventricular (AV) nodal conduction. Quinidine prolongs repolarization, an effect that is more prominent at slow heart rates (reverse use dependence) because of block of IKr (as well as enhancing the late Na current). Faster rates result in more block of sodium channels and less unblocking because of a smaller percentage of time spent in a polarized state (use dependence). Isoproterenol can modulate the effects of quinidine on reentrant circuits in humans. Quinidine at higher doses inhibits the late Na current. As noted, quinidine blocks the transient outward current Ito, which is probably why it is effective in suppressing ventricular arrhythmias in Brugada syndrome (see Chapter 9).
TABLE 35-5
In Vivo Electrophysiological Characteristics of Antiarrhythmic Drugs
| DRUG | ELECTROCARDIOGRAPHIC MEASUREMENTS | ELECTROPHYSIOLOGIC MEASUREMENTS | |||||||||
| Sinus Rate | PR | QRS | QT | JT | ERP-AVN | ERP-HPS | ERP-A | ERP-V | AH | HV | |
| Quinidine | 0 ↑ | ↓ 0 ↑ | ↑ | ↑ | ↑ | 0 ↑ | ↑ | ↑ | ↑ | 0 ↓ | ↑ |
| Procainamide | 0 | 0 ↑ | ↑ | ↑ | ↑ | 0 ↑ | ↑ | ↑ | ↑ | 0 ↑ | ↑ |
| Disopyramide | ↓ 0 ↑ | ↓ 0 ↑ | ↑ | ↑ | ↑ | ↑ 0 | ↑ | ↑ | ↑ | ↓ 0 ↑ | ↑ |
| Ajmaline | 0 | 0 ↑ | ↑ | ↑ | ↑ | 0 | ↑ | ↑ | ↑ | ↓ 0 ↑ | ↑ |
| Lidocaine | 0 | 0 | 0 | 0 ↓ | ↓ | 0 ↑ | 0 ↑ | 0 | |||



