(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Recent studies indicate that mitochondrial function is altered in the insulin-resistant myocardium, and this contributes to complex metabolic problems, such as obesity and diabetes mellitus, factors of significant cardiovascular mortality. Mitochondrial dysfunction and uncoupling are induced by fatty acids leading to reduction of cardiac efficiency by limiting ATP production and increasing myocardial oxygen consumption. Insulin resistance is a major factor in the pathogenesis of type 2 diabetes in the elderly and age-related changes associated with increased fat accumulation and reduction in mitochondrial oxidative phosphorylation activity, indicating that an age-associated decline in mitochondrial function contributes to insulin resistance in the elderly.
Introduction
Insulin maintains glucose homeostasis by stimulating glucose uptake in skeletal and cardiac muscles and suppressing production of glucose in the liver. Decreased sensitivity to these metabolic actions of insulin is a definition of “Insulin Resistance” (InsRes) [1]. This dysfunction is the principal feature of the cardiometabolic syndrome, a precursor to diabetes and subsequent cardiovascular complications.
There is good body of studies that has established the relation of insulin resistance to the development of type 2 diabetes mellitus and cardiovascular disease [2, 3]. For instance, nondiabetic, nonobese persons with hyperinsulinemia and impaired glucose tolerance compared to normoinsulinemic subjects showed a cluster of risk factors (e.g., elevated triglyceride and low-density lipoprotein levels, elevated systolic blood pressure) for coronary artery disease (CAD) [4]. The strongest relations between insulin resistance and CAD are seen in middle-aged persons and at higher elevations of plasma insulin levels [3]. When clustered with several other metabolic abnormalities (central obesity, hypertension, decreased high-density lipoprotein, elevated serum triglycerides), insulin resistance becomes a component of the metabolic syndrome (MS) which predicts the development of diabetes and cardiovascular disease.
MS has become a worldwide epidemic and a major public health-care concern. It is considered to be the cause of the current epidemic of diabetes and cardiovascular diseases (CVDs) [5]. MS occurs very frequently in the general population aged 40–79 years, and the identification of individuals with MS is useful from a clinical standpoint, as these individuals should benefit from interventions aimed at reducing their cardiovascular risk [6].
Mitochondria play an important role in energy homeostasis by metabolizing nutrients and producing energy in the form of ATP and heat, so the metabolic regulation depends largely on this organelle. Both mitochondrial function and insulin sensitivity are affected by genetic and environmental factors (the latter includes diet, aging, and stress). Importantly, it has been shown that mitochondrial dysfunction is associated with insulin resistance in skeletal muscle as well as in other tissues, including heart and vessels [7, 8].
Although fatty acids (FAs) serve as the chief energy substrate for the heart, the heart is capable of generating ATP from other substrates. Flexibility in shifts between oxidation of FAs and glucose to generate ATP provides the heart with an efficient and continuous fuel source in various physiological and nutritional conditions. Altered insulin signaling in the insulin-resistant heart reduces myocardial glucose uptake and utilization and increases uptake and utilization of FAs to meet cardiac energy demands [9, 10]. This shift in the spectrum of myocardial fuel preference serves initially as an adaptive function and involves mitochondrial biogenesis through activation of PGC-1α gene expression by the FA-activated nuclear receptor PPARα [11].
Unfortunately, long-term dependence on mitochondrial FA oxidation (FAO) creates an imbalance between energy intake and outflow leading to mitochondrial dysfunction, which is characterized by a reduced ratio of energy (ATP) production to respiration. Dysfunctional mitochondria stress the cardiomyocyte via accumulation of toxic lipid intermediates and reactive oxygen species (ROS), and increased myocardial oxygen consumption, which predisposes to cardiac dysfunction [12, 13]. In this chapter, we will discuss how insulin resistance-related mitochondrial dysfunction may be a contributory factor for many chronic diseases, in particular CVDs.
Mechanistic Development of Insulin Resistance: The Role of Mitochondria
Major actions of insulin in the heart include the regulation of glucose oxidation via stimulation of glucose transporter translocation and the regulation of protein synthesis. Also, insulin plays an essential role in the regulation of cardiac autophagy [14]. The biological actions of insulin are mediated by an insulin receptor (IR). IR consists of two α-subunits and two β-subunits, and has intrinsicligand-activated tyrosine kinase activity. Active IR phosphorylates intracellular substrates, IR substrate (IRS) proteins, and Shc, which serve as a scaffold for downstream signaling molecules [15, 16].
One signaling cascade triggered by insulin-activated IR involves IRS adaptor proteins. Tyrosine phosphorylation of IRS at multiple sites creates Src homology 2 (SH2)-domain-binding motifs—docking sites for SH2-domain-containing phosphatidylinositol 3-kinase (PI3K). PI3K is a heterodimer composed of a regulatory and a catalytic subunit. Binding of SH2 domains of PI3K regulatory subunit to IRS allosterically activates the catalytic subunit of PI3K, and kinase generates the lipid product phosphatidylinositol 3,4,5-trisphosphate (PIP3) from the substrate, phosphatidylinositol 4,5-bisphosphate (PIP2). PIP3 serves as an activator of 3-phosphoinositide-dependent protein kinase 1 (PDK1), and activated PDK-1 subsequently phosphorylates several downstream serine–threonine kinases (Akt, atypical protein kinase C isoforms). PI3K–PDK1–Akt-dependent branch of the insulin-signaling pathway is responsible for regulation of glucose uptake by glucose transporter GLUT4 (Fig. 21.1); regulation of physiological growth of cardiomyocytes by activation of mammalian target of rapamycin (mTOR)-dependent pathway and suppression of cell atrophy programs via inhibition of GSK3β and transcription factor Forkhead box O (FOXO).
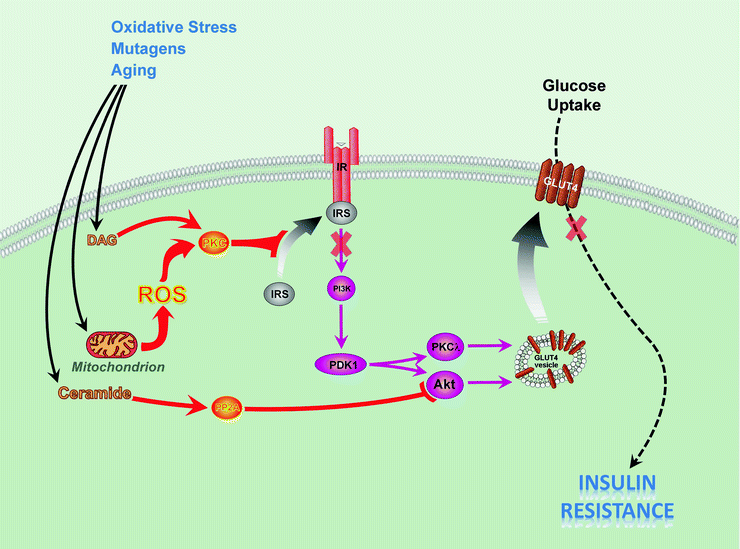
Fig. 21.1
Mechanisms of insulin resistance caused by mitochondrial dysfunction. Oxidative stress, mutations, and aging contribute to mitochondrial dysfunction and accumulation of diacylglycerol (DAG) and ceramide. Accumulation of DAG activates protein kinases C β, δ, θ (PKC). Mitochondrial dysfunction increases production of reactive oxygen species (ROS), which causes activation PKCs, serine phosphorylation of insulin receptor substrate proteins 1/2 (IRS), and subsequently results in insulin resistance. Increased serine phosphorylation of IRS-1/2 inhibits its interaction with insulin receptor (IR) and leads to decreased activity of insulin downstream signaling pathway, including phosphatidylinositol 3-kinase (PI3K), 3-phosphoinositide-dependent protein kinase 1 (PDK1), protein kinase B (Akt), and protein kinase C λ (PKCλ). Ceramide activates protein phosphatase 2A (PP2A) to remove activating phosphates from Akt. All abovementioned events culminate in decreased translocation of the glucose transporter 4 (GLUT4) to the plasma membrane and decreased glucose uptake in heart, skeletal muscle, and adipose tissue. The reduced insulin responsiveness (insulin resistance) causes diabetes and cardiovascular diseases
The starting point of another signaling pathway is IR-dependent phosphorylation of adaptor protein Shc. SH2-domain-binding motif on phosphorylated Shc attracts growth factor receptor-bound protein-2 (Grb-2) with preassociated GTP exchange factor Sos. Interaction with Shc and Grb-2 activates Sos, and it facilitates formation of active GTP-containing small GTP-binding protein, Ras, which then triggers a signaling cascade involving Raf kinase, MAP kinase kinase (MKK1), and the extracellular regulated kinases 1 and 2 (Erk1/2). This branch of insulin signaling regulates cell growth, mitogenesis, and differentiation.
InsRes has been suggested to develop as a result of defects at multiple sites in insulin-signaling pathway. One possible mechanism includes reduced tyrosine phosphorylation of IRS and subsequent decrease of PI3K activation. This can happen when activated serine–threonine kinases phosphorylate IRS proteins at serine sites that are known to inhibit their interaction with and tyrosine phosphorylation by IR [17, 18] (Fig. 21.1).
Dysfuntional mitochondria may contribute to the development of insulin resistance in several ways. First, abnormal functioning of mitochondria in the cells subjected to oxidative stress or in the cells with altered metabolism (see sections below) increases mitochondria-produced ROS which, in turn, activate various serine kinases that phosphorylate IRS proteins, leading to insulin resistance [19]. For instance, ROS stimulate IκB kinase beta (IKKβ) that phosphorylates IRS-1 at the serine residues [20]. At present, the detailed mechanisms for serine kinase activation mediated by ROS are not clearly understood. Under certain conditions (aging, mutagenic stressors—see sections below), the mitochondria become implicated in the accumulation of fatty acid metabolites, diacylglycerol (DAG), and long-chain fatty acyl-CoA (LCFA-CoA) [21]. DAG activates PKCs, including PKCβ, δ, and θ, that increase serine phosphorylation of IRS proteins, leading to inhibition of insulin signaling and insulin resistance [21–23] (Fig. 21.1). The PKCθ-deficient mouse is protected from fat-induced InsRes [24].
Oxidative Stress and Mitochondrial Function
The heart possesses a relatively low endogenous antioxidant capacity that is contributed by both enzymatic and nonenzymatic free radical scavengers and antioxidants, making it susceptible to oxidative stress with attendant structural and functional abnormalities [25].
In the rodent model of InsRes, which develops in response to inappropriate activation of the renin–angiotensin–aldosterone system (RAAS) (transgenic Ren2 rats), InsRes has been observed associated with oxidative stress [26–28]. In particular, enhanced RAAS culminates in elevated tissue levels of angiotensin II (Ang II). Ang II induces NADPH oxidase leading to overproduction of extramitochondrial ROS and oxidative stress. There is emerging evidence that NADPH oxidase-derived extramitochondrial ROS can promote mitochondrial oxidative stress [29, 30] associated with increased generation of mitochondrial ROS.
In InsRes, the excess of ROS generated by mitochondria contributes to downregulation of genes required for mitochondrial oxidative phosphorylation [31, 32]; to protein, DNA, and lipid injury; and, ultimately, to reduction in mitochondrial biogenesis and mitochondrial dysfunction [31, 33]. Also, increases in ROS, in turn, activate various serine kinases that phosphorylate IRS proteins, leading to further development of InsRes [19] (Fig. 21.1). On the other hand, defects in the antioxidant capacity of ventricular cardiomyocytes may be involved in the cardiac remodeling and hypertrophy caused by InsRes in high-fat diet rodents [32].
Insulin Resistance and Mitochondrial Biogenesis
Studies on mitochondria indicate that diabetes is associated with mitochondrial abnormalities in both skeletal muscle and heart, for instance, mitochondrial structural and functional derangements have been shown in skeletal muscle of insulin-resistant and diabetic animal models and humans [19, 34–37]. Similarly, diabetes-related mitochondrial abnormalities are demonstrated in the heart [38, 39]. At the same time, the role of the mitochondrial abnormalities as causal or secondary in diabetic cardiac dysfunction is still questionable. The study from Duncan et al. [11] has shed some light to this dilemma. According to these investigators, functional alterations in mitochondria start in the prediabetic, insulin-resistant stage, possibly as an adaptive response to support increased flux of fatty acids through the mitochondrial β-oxidation pathway (Fig. 21.2): in the hearts of insulin-resistant uncoupling protein-diphtheria toxin A (UCP-DTA) transgenic mice, fatty acid-activated nuclear receptor peroxisome proliferator-activated receptor-α (PPARα), possibly together with the transcriptional coactivator, PPARγ coactivator-1α (PGC-1α), activates the expression of nuclear and mitochondrial genes encoding enzymes that are involved in multiple mitochondrial pathways. Duncan et al. [11] results demonstrated that mitochondrial biogenesis occurs early in the development of diabetic cardiac dysfunction through a transcriptional regulatory circuit that involves activation of PGC-1α gene expression by the fatty acid-activated nuclear receptor PPARα.
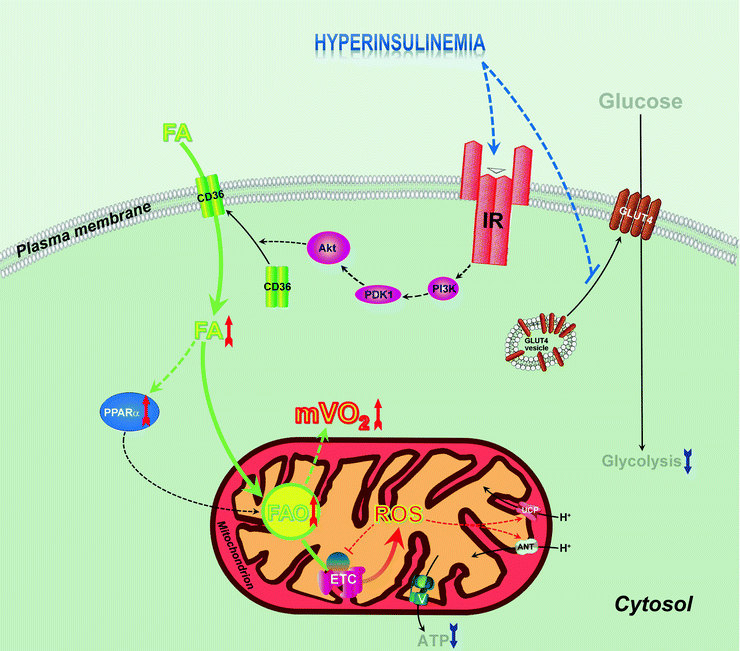
Fig. 21.2
Mitochondrial dysfunction in insulin-resistant states. Hyperinsulinemia activates insulin receptor (IR)-dependent pathway involving phosphatidylinositol 3-kinase (PI3K), 3-phosphoinositide-dependent protein kinase 1 (PDK1), and protein kinase B (Akt1), resulting in increased translocation of CD36/FATP transporter to myocardial plasma membrane, which leads to increased fatty acid (FA) uptake. Insulin-resistant state is also characterized by decreased translocation of glucose transporter 4 (GLUT4) leading to decreased glucose uptake and decreased glycolysis, which further increases FA utilization. Increased FAs activate peroxisome proliferator-activated receptor alpha (PPARα), positive regulator of expression of proteins involved in FA utilization, and this results in increase of FA β-oxidation (FAO). Increased FAO is associated with increased myocardial oxygen consumption (mVO2). Progression of insulin resistance is associated with overproduction of reactive oxygen species (ROS) in mitochondria which facilitate mitochondrial uncoupling via stimulation of uncoupling proteins 2 and 3 (UCP) and adenine nucleotide translocator (ANT) leading to further increase of mVO2. ROS-dependent damage of components of electron transport chain (ETC) decreases ATP generation by ATP synthase (V) and decreases cardiac efficiency
First, in the insulin-resistant heart, increased circulating FAs activate PPARα/PGC-1α to induce FA uptake and oxidation (Fig. 21.2). Also, PPARα/PGC-1α accelerate mitochondrial biogenesis characterized by an increase in mitochondrial volume density and mtDNA content. Biogenesis of mitochondria in insulin-resistant myocardium seems to play a compensatory role to increase respiratory capacity of mitochondria inefficient under these conditions [40].
Factors that regulate expression of PGC-1 influence insulin resistance. Thus, endothelial nitric oxide synthase (eNOS) regulates PGC-1 expression via eNOS–NO–cGMP pathway and is involved in mitochondria biogenesis [8, 41]. eNOS-deficient mice are insulin resistant and have defects in fatty acid metabolism and fewer mitochondria [41–43]. Another regulator of PGC-1α and promoter of mitochondrial biogenesis is AMP-activated protein kinase (AMPK) [44–46]. AMPK activates PGC-1 by direct phosphorylation on threonine and serine residues, and this covalent modification stimulates mitochondrial biogenesis [47, 48].
As mentioned earlier, a series of events in insulin-resistant myocardium is triggered by PPARα/PGC-1α as an adaptive response to increase capacity for mitochondrial FAO and manage mitochondrial inefficiency. Unfortunately, despite an increasing in mass, mitochondria under InsRes, obesity or type 2 diabetes mellitus conditions exhibit significant decrease of oxidative phosphorylation and reduced expression of complex I, III, and V of the electron transport chain (ETC). Expression of many mitochondrial OXPHOS genes and mitochondrial transcription factor A (TFAM) is regulated by nuclear transcription factor, nuclear respiratory factor-1 (NRF-1), and its coactivator, PGC-1 [49]. Compared to normal humans, levels of PGC-1 can decrease in InsRes and in diabetic subjects, while levels of NRF-1 are reduced in the diabetic [36]; these changes probably underlie a coordinated downregulation of genes of oxidative metabolism in humans with InsRes and diabetes, which have been reported in several studies [36, 50]. It is worthwhile to note that levels of expression of PGC-1α, an NRF, do not always fully account for mitochondrial dysfunction in insulin-resistant patients. For instance, Morino et al. [19] reported that the mitochondrial function was significantly decreased in insulin-resistant offspring of type 2 diabetes mellitus parents compared to control subjects, but levels of expression PGC-1α, NRFs, and TFAM were not different.
Aging and Mitochondrial Function
Aging may be associated with insulin resistance, which often is attributed to obesity and physical inactivity [51–53]. It has been documented that InsRes in aging skeletal muscle and β-cells is associated with mitochondrial alterations [54–56], although only recently the role of dysfunctional mitochondria in the development of insulin resistance in the aged myocardium has been demonstrated. Bhashyam et al. [57] discovered that in large-animal model of aging (senescent beagles), old dogs have impaired insulin-stimulated myocardial glucose uptake, which is associated with altered insulin signaling and glucose transporter GLUT4 translocation. Importantly, myocardial insulin resistance in this animal model is independent of obesity and physical inactivity.
Altered myocardial insulin-mediated glucose uptake in older dogs is associated with increased fatty acid uptake, lipid accumulation, and myocardial oxygen consumption which are probably related to a reduced expression of mitochondrial uncoupling protein (UCP) 3 and several components of ETC (mitochondrial cytochrome c oxidase 1, succinate dehydrogenase A). Interestingly, insulin resistance is associated with impaired insulin-induced Akt1 phosphorylation and GLUT4 translocation in both cardiac and skeletal muscles, but the mechanism is different. Thus, decrease in insulin-dependent phosphorylation of Akt1 in myocardium is based on reduction in IR density and increased expression of protein tyrosine phosphatase (phosphatase and tensin homolog, PTEN), whereas in skeletal muscle decreased insulin–IR–Akt1 signaling involves serine phosphorylation of IRS-1 [57].
Impaired eNOS signaling contributes to aging-associated myocardial insulin resistance and mitochondrial function. Li et al. [58] demonstrated that exercise (swim training) upregulates eNOS expression in aging rat heart, improves myocardial insulin-dependent glucose uptake, and facilitates mitochondrial function. Exercise-improved myocardial insulin sensitivity and mitochondrial insulin-induced O2 consumption were abolished by an eNOS inhibitor, cavtratin.
Although there is no literature on the myocardium, possible role of mitochondrial dysfunction in insulin resistance was demonstrated in human aging skeletal muscle [34, 56, 59]. Healthy elderly human subjects have ∼40% reduction in skeletal muscle oxidative phosphorylation as a result of reduction in mitochondrial number [56, 60, 61] and accumulation of mutations in control sites for mitochondrial DNA replication [62]. Defects in mitochondrial oxidative and phosphorylation capacity lead to increases in intramyocellular fatty acid metabolites [56] and to the increased triglyceride content in muscle [59]—factors related to the development of insulin resistance in the elderly. Also decrease of PGC-1 expression [63], reduction of AMP-activated protein kinase (AMPK) activation [47], and alteration of genes related to fatty acid oxidation [61, 64] may contribute to decreased mitochondrial biogenesis and insulin resistance accompanying the aging process in skeletal muscle.
It should be mentioned here that there are substantial differences in age-related changes of cardiac functional and metabolic responses to insulin between different species. Thus, in contrast to dog and rat, the aged murine heart exhibits substantial metabolic plasticity and maintains cardiac function by recruiting and oxidizing alternative substrates (glucose, acetoacetate, lactate) [65]. Moreover, fatty acid uptake is markedly impaired in aged mouse heart due to reductions in protein expression for PPARα, a transcriptional activator of pyruvate dehydrogenase kinase 4. Also, insulin does stimulate an inotropic response in aged mouse myocardium, supported by increased oxidation of glycolytic substrate, but further suppresses FA oxidation [65].
Genetic Factors and Mitochondrial Function
As discussed in the section on “Mechanistic Development of Insulin Resistance,” dysfunctional mitochondria play a role in the development of InsRes in several tissues including myocardium. Genetic factors contribute to mitochondrial dysfunction and InsRes through polymorphism of nuclear and mitochondrial genes encoding mitochondrial proteins, ribosomal and transfer RNAs, which are all necessary for the normal functioning of mitochondria.
Single-nucleotide polymorphism (an A3243G substitution) in the mitochondrial transfer RNA (tRNA)Leu(UUR) gene is associated with impaired insulin sensitivity in about 1.5% of the diabetic population in different countries and races [66, 67]. The underlying mechanism of this pathology involves a delayed insulin secretion due to an impaired mitochondrial ATP production in consequence of the mtDNA defect [68], OXPHOS dysfunction in the skeletal muscle (defective cytochrome c oxidase subunits II and III, defective subunit 6 of ATP synthase) [69, 70], and an isolated respiratory complex I defect in heart and skeletal muscle [71]. Similarly, T3271C transversion in tRNALeu(UUR) also causes a moderate mitochondrial translation defect, decreased synthesis of subunit 6 of respiratory complex I (ND6), and complex I deficiency [72]. Several other polymorphic variants of mitochondrial tRNA genes also demonstrate insulin-linked pathology phenotype in association with mitochondrial cytochrome c oxidase deficiency: A7472C-tRNASer(UCN) and C12258A-tRNASer(AGY) genes (in skeletal muscle and the left ventricle biopsies) [73, 74], heteroplasmic A8296G mutation in tRNALys gene (various tissues of patients including heart) [75], and T14709C mutation in tRNAGlu gene (patient’s muscle) [76]. Thus, amino acid misincorporation might be a common theme to mutations in mitochondrial tRNAs that are associated with the development of insulin-related abnormalities through functional impairment of mitochondria.
The mitochondrial genome is very susceptible to various mutagenic stressors because mitochondrial genes are not protected by histones and they are positioned closely to ETC, which produces ROS [77], and mammalian mitochondrial DNA is known to develop mutations with aging [77–81]. Aberrant expression and improper functioning of mutant mitochondrial OXPHOS genes are related to mitochondrial dysfunction, and increased insulin resistance and non-insulin-dependent diabetes mellitus in the elderly [56, 78]. Analogously, ROS induce mtDNA mutations that lead to the synthesis of functionally impaired respiratory chain subunits, causing respiratory chain dysfunction [82].
Nuclear-encoded protein components of mitochondrial oxidative metabolism are also involved in the development of InsRes. For example, insulin resistance can be associated with low expression of nuclear-encoded mitochondrial genes (such as subunit NDUFA3 of complex I, subunit VIc of cytochrome c oxidase) because of reduced level of nuclear regulator PGC-1α [36, 50, 83, 84]. Muller et al. reported that altered lipid oxidation and early insulin secretion in Pima Indians is associated with Gly482Ser missense mutation in this regulator [85]. Another genetic disorder is caused by defects in nuclear-encoded acyl-coenzyme A dehydrogenase 9 and affects mitochondrial fatty acid β-oxidation [86].
Interestingly, experimentally induced mutations of certain genes in animal models uncover the roles of those genes in the functioning of mitochondria under insulin resistance conditions. For instance, eNOS-deficient insulin-resistant mice demonstrate better survival after pressure-induced dysfunction of myocardium than eNOS-expressing insulin-resistant animals. Probably in conditions of insulin resistance, eNOS increases oxidative stress and exacerbates ventricular remodeling via influence mitochondria [87].
Cardiovascular Metabolic Syndrome: Mitochondrial Bioenergetics and Biogenesis Defects
Metabolic syndrome [also known as metabolic syndrome X, cardiometabolic syndrome, syndrome X, insulin resistance syndrome, Reaven’s syndrome (named for Gerald Reaven)] is a combination of medical disorders that, when occurring together, increase the risk of developing cardiovascular disease and diabetes. These disorders include insulin resistance/hyperglycemia, obesity/dyslipidemia (elevated triglycerides, reduced HDL cholesterol), and hypertension [88–92] (Fig. 21.3).

Fig. 21.3
Metabolic syndrome and associated complications: role of mitochondria. In metabolic syndrome-associated complications, cardiac mitochondria generate excess of reactive oxygen species (ROS) as a consequence of elevated accumulation/β-oxidation of free fatty acids (FAO). ROS negatively influence components of mitochondrial oxidative phosphorylation machinery (OXPHOS) and damage mitochondrial DNA (mtDNA) leading to reduced mitochondrial function and further progression of metabolic abnormalities. Eventually, it may result in the development of cardiovascular diseases. IR insulin resistance
It is well documented that metabolic syndrome, associated with obesity, InsRes, and hypertension, is accompanied by impaired myocardial diastolic relaxation (diastolic dysfunction)—a reduction in velocity of myocardial relaxation as well as decreasing myocardial compliance [93–95]. Mechanism that contributes to this cardiac dysfunction involves malfunction of mitochondria: decrease in energy production due to reduced mitochondrial respiration and increased oxidative stress [96, 97]. In obese, InsRes persons as well as diabetic patients, these changes are associated with reductions in the oxidative capacity of the mitochondrial ETC [31, 97]. Changes in mitochondrial biogenesis in the heart have also been documented in the metabolic syndrome and diabetes [8, 98].
In the rodent model of InsRes, ultrastructure analysis of myocardium supports myocardial mitochondrial biogenesis—increased numbers of smaller abnormal mitochondria [28]. Оveractive renin–angiotensin–aldosterone system (RAAS) and associated increases in oxidative stress play an important role in the development of mitochondrial abnormalities in metabolic-related cardiomyopathies in the metabolic syndrome. For instance, inappropriate activation of the RAAS in the transgenic Ren2 rat (rodent model of InsRes demonstrating symptoms of metabolic syndrome) is accompanied by increased Ang II and ROS in the heart [27, 28]. ROS can cause damage of mitochondrial DNA (large deletions) which enhances the biogenesis of cellular mitochondria [31]. Thus, increased mitochondrial biogenesis in InsRes models is an adaptive process to generate ATP in response to enhanced myocardial energy requirements. However, this leads to increased numbers of small abnormal, less efficient mitochondria. Importantly, inhibition of the RAAS leads to attenuation of mitochondrial abnormalities in transgenic Ren2 rat [27, 28].
As we already mentioned earlier, mitochondrial biogenesis is driven, in part, through PGC-1α, an integrator of the transcriptional network regulating mitochondrial biogenesis (see previous sections for the role of PGC-1α in the mediation of NRFs which, in turn, govern downstream genes including TFAM and OXPHOS components). Individuals with InsRes display fewer, and sometimes smaller, skeletal muscle mitochondria, which could be attributed to reduced PGC-1α and PGC-1β expression [49, 99]. Thus, accumulating data support the notion that dysregulation of PGC-1α may lead to an abnormal mitochondrial function and mitochondrial density associated with the metabolic pathology (InsRes).
While contribution of InsRes to mitochondrial dysfunction is well validated [100] (and also see previous sections of this chapter), less is known regarding the role of other metabolic risks in development of mitochondrial dysfunction and metabolic cardiomyopathy. In a very few studies that have attempted to examine mitochondrial function in the human heart in obesity and type 2 diabetes, the obesity has been associated with an increase in myocardial oxygen consumption, whereas impaired glucose tolerance correlated with increased FA utilization [101–103]. This suggests that a perturbation of mitochondrial energy metabolism may contribute to the impaired cardiac contractility observed in obese subjects. The increased myocardial fatty acid oxidative capacity in obesity and diabetes are mediated, in part, by increased activity of PPARα, a central regulator of FA oxidation in the heart, and increased expression of PGC-1α, a potent transcriptional coactivator of PPARα [104]. An increased PPARα signaling may sustain elevated rates of fatty acid oxidation in the heart as diabetes persists.
Recently, failure of DNA repair was proposed to generate defects in cell proliferation, apoptosis, and mitochondrial dysfunction which in turn promotes atherosclerosis and the development of metabolic syndrome. In particular, Mercer et al. [105] demonstrated that haploinsufficiency in DNA repair coordinating protein, ataxia telangiectasia mutated kinase (ATM), accelerates atherosclerosis and induce multiple features of the metabolic syndrome (hypertension, hypercholesterolemia, obesity, steatohepatitis, glucose intolerance) in rodent model of atherosclerosis (apolipoprotein E knockout mice). ATM-insufficient animals show increased nuclear and mitochondrial DNA damage, reduced mitochondrial oxidative phosphorylation, and induced metabolic changes consistent with mitochondrial defects (increased β-hydroxybutyrate, reduced lactate, reduced glucose, and alterations in multiple lipid species).
In summary, persistent DNA damage in plaque cells of ATM-deficient animals changes the rates of cell proliferation and cell death that might directly promote atherosclerosis. Moreover, insufficient levels of DNA repairing protein promote mitochondrial dysfunction via increased ROS, mtDNA damage, and mtDNA deletion. Reduced complex I activity and reduced oxidative phosphorylation in impaired mitochondria further increase ROS and mitochondrial dysfunction, lead to lipid accumulation and glucose intolerance, and finally promote atherosclerosis and features of the metabolic syndrome [105] (Fig. 21.3).
Growing evidence indicates that oxidative stress and chronic inflammation are initiating events in the development of atherosclerosis which is common for type 2 diabetes, metabolic syndrome, and cardiovascular disease, with the role of endothelial cell dysfunction as an early step. According to the “fetal programming hypothesis” [106], endothelial cell dysfunction may occur in growth-restricted neonates exposed to placental insufficiency and might be involved in the occurrence of atherosclerosis later, in adulthood. Several lines of evidence indicate that mitochondrial damage is central to this process: elevated ROS production by mitochondria when chronically exposed in utero to oxidative stress and inflammation—for instance, in pregnancies complicated by preeclampsia—damages lipids, proteins, and mtDNA leading to mitochondrial dysfunction [107] with subsequent endothelial cell dysfunction, vascular smooth muscle cell proliferation, and apoptosis resulting in the development of atherosclerosis [108].
Conclusions
Insulin resistance plays a central role in the pathogenesis of the cardiometabolic syndrome, type 2 diabetes mellitus, and subsequent cardiovascular complications. An early and consistent finding in InsRes is an increase in myocardial fatty acid oxidation. The mechanisms for the increase in fatty acid utilization are multifactorial; recent studies have shown an important role for changes in glucose utilization as a potential initial inciting event that leads to increased oxidation of fatty acids. Later changes are driven by increased FA-dependent activation of PPARα targets. Increased fatty acid oxidation is associated with decreased cardiac efficiency, which is in part the result of the increased oxygen costs of fatty acid oxidation, with contributions from mitochondrial uncoupling in insulin-resistant models with diabetes and impaired glucose tolerance.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


