The Ischemic Heart
The heart requires an uninterrupted supply of oxygen to meet its high energy needs, which can be satisfied only by oxidative metabolism (Chapter 2). Coronary artery occlusion causes an almost immediate loss of function and, within hours, death of the energy-starved cardiac myocytes. Ischemic myocardial cell death, called myocardial infarction, is a major public health problem in developed countries. Although this condition is often referred to as ischemic heart disease, the cause lies in the coronary arteries, so that the myocardium is the victim and not the perpetrator. For this reason, the alternative names coronary heart disease, arteriosclerotic heart disease, and atherosclerotic heart disease are commonly used.
Conditions other than coronary occlusion can cause the heart to become energy-starved. These include aortic stenosis and pulmonary hypertension, in which pressure overload increases oxygen requirements to levels that exceed the amount that can be supplied by even a normal coronary circulation. Severe anemia, by reducing the oxygen-carrying capacity of the blood, also causes the heart to become energy-starved. Angina pectoris (literally strangling in the chest), the typical symptom when the heart’s demand for oxygen exceeds the amount supplied by the coronary circulation, is therefore not diagnostic of coronary heart disease.
Coronary Occlusive Disease
Coronary atherosclerosis, by far the most common cause of coronary occlusion and myocardial infarction, is the result of a chronic inflammatory process that evolves over many decades. A sudden decrease in coronary flow usually occurs when endothelial damage leads to the formation of platelet plugs and thrombi (clots) that occlude one or more major coronary arteries. These occlusive events, which generally evolve over periods of hours or days, are most commonly initiated by disruption of a fibrous cap that separates soft lipid-filled atherosclerotic lesions within the arterial wall from the lumen. This process, called plaque rupture, exposes collagen and other thrombogenic elements in the atherosclerotic lesion to the moving column of blood in the coronary artery, which triggers platelet aggregation and thrombus formation that occlude the diseased vessel. Vasospasm, a less common cause of coronary occlusion, can be caused when vasoconstrictor peptides are released from activated platelets in diseased arteries that may lack obvious atherosclerotic lesions.
Total occlusion of a major coronary artery generally causes the entire thickness of the ventricular wall to become ischemic, a condition called transmural ischemia. However, in a patient with well-developed coronary artery collaterals, a similar occlusion may cause significant ischemia only in the endocardium, where the balance between energy supply and energy demand is most precarious. This is because, compared to the epicardium, the endocardium is less well perfused (see Chapter 1) and its wall stress is higher (see Chapter 11). Increased myocardial oxygen
demand in a patient with a partial coronary artery occlusion can also cause the endocardium to become ischemic; this is the basis for the ECG evidence of subendocardial ischemia in a positive stress test (see later).
demand in a patient with a partial coronary artery occlusion can also cause the endocardium to become ischemic; this is the basis for the ECG evidence of subendocardial ischemia in a positive stress test (see later).
Angina Pectoris
Angina pectoris, the typical symptom caused when cardiac energy demand exceeds energy supply, is a visceral discomfort that localizes poorly and is often difficult to describe. Most patients experience a heaviness or squeezing in the left chest, but others note only a vague discomfort that may be mistaken for indigestion. Angina typically radiates to the inner left arm, neck, or jaw, but may be referred elsewhere; for example, to the site of an old back or shoulder injury or the socket of a lost tooth. Not all patients experience typical symptoms, even when total coronary occlusion has caused a large myocardial infarction. The substance or substances responsible for the chest discomfort remain unclear.
The symptoms of ischemic heart disease appear in patterns that depend on how they are provoked. Demand angina, which as the name implies occurs when the symptoms are provoked by increased cardiac work, as occurs during exercise, almost always forces patients to stop exertion and is typically relieved promptly by rest, usually within a few minutes. The severity of demand angina can be quantified in terms of the amount of effort needed to provoke symptoms, most precisely by a stress test. In supply angina, which typically occurs when the patient is at rest, the imbalance between energy supply and energy demand is caused by a decrease in supply, so that this pattern indicates that coronary flow has been reduced by a new occlusion or vasospasm.
One of the most important characteristics of angina pectoris is its stability; that is, whether the discomfort is staying the same, improving, or worsening. Stable angina can remain unchanged for years because coronary atherosclerosis progresses slowly, often in a stepwise manner. Spontaneous improvement in the symptoms of cardiac ischemia, which was common before procedures were developed to revascularize the heart, occurs when blood flow to underperfused regions is increased by enlargement of partially occluded coronary arteries, called vascular remodeling, and by the development of collateral vessels. Unstable angina, where the symptom increases in severity, lasts longer, or is provoked by decreasing effort, indicates that the underlying coronary disease has entered a dangerous period when acceleration of the underlying vascular disease has increased the risk for a myocardial infarction and sudden death.
Stable Angina
Symptoms in patients with stable angina pectoris typically follow the pattern of demand angina, in which the chest discomfort is exacerbated by increased myocardial oxygen demand, for example, exertion or emotional upset. Stable angina generally has a benign prognosis because the brief episodes of ischemia do not damage the heart, but instead increase the ability of the myocardium to withstand a fall in blood flow (see later). For this reason, the natural history of stable angina is determined largely by progression of the disease in the coronary arteries. The key to improving prognosis in these patients is meticulous management of the risk factors for atherosclerosis that include high cholesterol, diabetes, hypertension, obesity, and elimination of smoking. When symptoms become troublesome, patients are usually treated with β-adrenergic blockers and arteriolar vasodilators to reduce the heart’s energy demand, nitrates which decrease preload and dilate coronary collateral vessels, and drugs that
inhibit platelet aggregation and clotting in the diseased coronary arteries. Revascularization by coronary artery bypass surgery or angioplasty usually alleviates symptoms but has little or no ability to prolong survival.
inhibit platelet aggregation and clotting in the diseased coronary arteries. Revascularization by coronary artery bypass surgery or angioplasty usually alleviates symptoms but has little or no ability to prolong survival.
Vasospastic Angina
Patients with coronary vasospasm typically experience angina when they are at rest, usually without a clear predisposing cause. This syndrome, which is also called vasospastic angina, variant angina, or Prinzmetal’s angina, is dangerous because, unlike demand angina in which myocardial energy starvation can be alleviated by rest, ischemia caused by vasospasm persists until the constricted coronary artery dilates. In some patients, myocardial ischemia lasts long enough to cause a myocardial infarction; in others, coronary vasospasm can trigger arrhythmias that may be fatal. The obvious goal of treatment is to relieve the coronary vasoconstriction, which can be done with variable success using combinations of nitrates, calcium channel blockers, and other vasodilators.
Acute Coronary Syndromes: Unstable Angina and Acute Myocardial Infarction
Worsening of angina, called acute coronary syndrome or instability, usually follows one of three patterns. The least severe, called unstable angina, occurs when the symptoms worsen and are provoked more easily, but there is no evidence of myocardial cell death. The other two, where increasing angina is accompanied by evidence of necrosis, are non–ST elevation myocardial infarction (NSTEMI) and ST elevation myocardial infarction (STEMI). These were formerly called subendocardial, non–Q-wave, or non-transmural myocardial infarction; and transmural or Q-wave myocardial infarction, respectively (see later). Although all acute coronary syndromes result from increasing coronary occlusion, they differ in pathophysiology and prognosis, and require different therapy.
Unstable Angina
The pathophysiology of angina and its relationship to energy starvation explains the clinical picture in patients with unstable angina; the intensity of the symptoms increases, the amount of effort needed to evoke the chest discomfort decreases, and angina can occur at rest. The appearance of unstable angina usually indicates that the coronary occlusive disease is worsening but can also be caused when a complicating condition, like anemia, impairs oxygen delivery to the heart. Unstable angina typically follows a “stuttering” course because platelet plugs and thrombi that form in the damaged artery break up or dissolve, and then reappear. The resulting impairment of blood flow is often exacerbated by coronary vasospasm caused by vasoconstrictor substances that are released when platelets are activated by the damaged endothelium.
Instability is generally initiated by disruption of an atherosclerotic plaque, where damage to the fibrous cap in a previously stable coronary artery lesion exposes a thrombogenic surface that leads to arterial occlusion. The appearance of unstable angina has ominous prognostic implications as it can herald a myocardial infarction or sudden cardiac death. For this reason, unstable angina requires prompt medical attention.
Acute Myocardial Infarction
Acute myocardial infarction occurs when coronary flow is reduced to levels so low as to cause myocardial cell death. Most clinical infarctions involve the left ventricle; right ventricular infarction can occur, but is less common. Because the endocardium is especially vulnerable to energy starvation (see earlier), coronary occlusion causes endocardial myocytes to be severely injured and to die sooner than those in the epicardium.
The criteria now used to classify patients with myocardial infarction depend on whether the ST segments in ECG leads facing the infarction are depressed or elevated. The distinction between STEMI and NSTEMI is determined largely by whether the full thickness of the ventricular wall or just the subendocardial layers are injured or infarcted (see later).
Prompt restoration of the normal balance between energy delivery and energy utilization is central to managing any of the acute coronary syndromes. Most important is to increase energy supply by addressing the underlying disease in the coronary arteries. This can be accomplished using aspirin and other platelet inhibitors, antithrombotic drugs, and thrombolytic agents. In patients with STEMI, the most effective treatment is reperfusion of the ischemic myocardium by immediate (primary) angioplasty. Reducing cardiac energy demands can also alleviate symptoms and improve prognosis, but this approach deals with the consequences, rather than the cause, of the clinical syndrome. β-Adrenergic blockers administered immediately after a coronary occlusion have important energy-sparing effects that are especially beneficial in regions where perfusion is reduced, but not totally abolished. Vasodilators, such as nitrates and calcium channel blockers, are useful because their ability to dilate arterioles that determine peripheral vascular resistance reduces afterload and so decreases energy demand. Nitrates also dilate veins, which has the added benefit of decreasing preload; these drugs also increase blood supply to ischemic regions of the heart by dilating intracoronary collateral vessels.
Jeopardized Myocardium and “Ischemia at a Distance”
The consequences of coronary occlusion are influenced by collateral vessels that, by connecting large epicardial coronary arteries, allow more than one major coronary artery to supply a given region of the heart (Fig. 17-1). Collateral vessels generally develop when partial occlusion of a coronary artery reduces, but does not totally interrupt, blood flow to a region of the heart. Because the collateral circulation increases with advancing age, occlusion of a major coronary artery in young patients, who typically have few collateral vessels, usually causes a large infarct, whereas the same occlusion in an older patient with an extensive collateral circulation may cause only minor symptoms, and sometimes no symptoms at all.
The role of collaterals can be understood by considering a patient with a large collateral vessel connecting a normal right coronary artery (RCA) to a partially occluded left anterior descending coronary artery (LAD), which together provide the anterior wall of the left ventricle with a dual blood supply (Fig. 17-1A). Occlusion of the RCA in this setting not only leads to necrosis of the inferior wall, which had received its entire blood supply from this vessel, but causes blood flow to the anterior wall to become jeopardized because the latter has become entirely dependent on the partially occluded LAD for its entire blood supply (Fig. 17-1B). Because the RCA occlusion has caused ischemia in a region (the anterior wall) that is outside of its normal distribution, this situation is called ischemia at a distance.
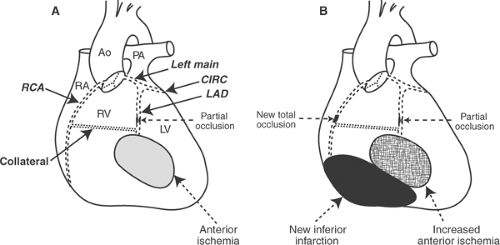 Fig. 17-1: Ischemia at a distance. A: Anterior view of a heart where partial occlusion of the LAD has stimulated the development of a collateral vessel linking a normal RCA to the occluded artery. The anterior region of the left ventricle supplied by the partially occluded LAD can become ischemic when the energy demands of the left ventricle are increased (light shading). B: A new total occlusion of the RCA causes infarction of the inferior region of the left ventricle to which it had provided the only source of arterial blood (black), and in addition decreases coronary flow to the anterior region that had previously received the dual blood supply (cross-hatched). The new RCA occlusion, therefore, reduces the blood supply to the jeopardized region that formerly had a dual blood supply, but which must now depend entirely on the partially occluded LAD. In this way, RCA occlusion caused ischemia at a distance because ischemia appeared in a region (the anterior wall) that is not normally supplied by the RCA. LAD: left anterior descending coronary artery; RCA, right coronary artery; CIRC: circumflex coronary artery; RA: right atrium; LA: left atrium; RV right ventricle; LV: left ventricle; PA: pulmonary artery; Ao: aorta. |
Initial Consequences of Coronary Artery Occlusion
Patients who sustain an acute myocardial infarction generally experience severe angina, loss of pump function, and arrhythmias. The initial hemodynamic consequences depend largely on the amount of the left ventricle that has been infarcted, but the severity of the arrhythmias and occurrence of sudden death in patients with an acute myocardial infarction do not correlate well with infarct size.
Two reflexes influence the clinical picture in acute myocardial infarction. The hemodynamic defense reaction (see Chapter 8), whose major effects in this setting are mediated largely by sympathetic stimulation, is activated because these patients are generally in pain and terrified; this response becomes even more powerful when left ventricular damage causes blood pressure to fall. Although the neurohumoral response helps maintain blood pressure, the ability of β-adrenergic stimulation to increase heart rate and contractility worsens energy starvation in the ischemic myocardium, as do arteriolar vasoconstriction and increased afterload caused by α-adrenergic stimulation. Sympathetic stimulation can also cause arrhythmias and sudden death. The second reflex that is activated in patients with inferior and posterior left ventricular infarction is the von Bezold–Jarisch reflex, a powerful vagal response that lowers blood pressure, slows the sinus node pacemaker, and can lead to AV block.
Hypoxia, like ischemia, causes energy starvation, but the consequences differ because coronary occlusion, in addition to interrupting oxygen supply to the heart, prevents the removal of metabolites and, by reducing the pressure within the coronary arteries supplying the left ventricle, attenuates the “garden hose effect” (see later).
Early Pump Failure
Interruption of coronary flow to the mammalian heart is followed within a few seconds by a decrease in contractility and profound impairment of ventricular filling (Fig. 17-2). The decrease in contractility, which is accompanied by abbreviation of systole, causes the ischemic region to bulge outward during systole because it cannot overcome the intraventricular pressure generated by the normally perfused myocardium. The resulting decrease in ejection also impairs filling because acute ventricular dilatation within the non-distensible pericardium reduces the ability of the heart to fill. This negative lusitropic effect, which is similar to pericardial tamponade, can be of greater hemodynamic importance than the negative inotropic effect.
The Garden Hose Effect
It was once believed that lack of adenosine triphosphate (ATP) was the major cause of the early pump failure that follows a coronary artery occlusion. However, the initial decrease in myocardial contractility precedes a significant fall in ATP content (Fig. 17-2). The most likely explanation for the rapid impairment in ejection is attenuation of the distending effect of intracoronary pressure (the garden hose effect, see Chapter 10).
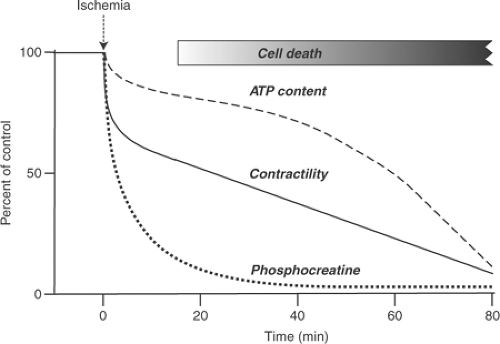 Fig. 17-2: Time courses of the decline in contractility, adenosine triphosphate content, and phosphocreatine after complete interruption of coronary flow. (Based on data from Williamson, 1966.) |
Metabolic Abnormalities
Oxygen tension within the myocardium falls almost to zero within a minute after complete cessation of blood flow. This reflects the very high affinity of the respiratory chain for oxygen, which allows the ischemic myocardial cells to consume all of the available oxygen within a few minutes after the myocardium loses its blood supply; as a result, oxidative phosphorylation comes to a complete halt. Anaerobic energy production is increased in part by decreases in ATP and glucose-6-phosphate levels, increased adenosine diphosphate (ADP), adenosine monophosphate (AMP), and Pi levels (see Chapter 2), and by the accelerated glycogen breakdown, glucose uptake, and glycolysis caused by sympathetic stimulation. Sympathetic stimulation also stimulates fatty acid release from triglycerides, but because these lipids cannot be oxidized, fatty acids and their derivatives accumulate in the ischemic heart (see later).
Although the rate of anaerobic glycolysis initially increases in the ischemic heart, this response cannot be sustained because NAD+ levels rapidly decrease to levels below those needed to reduce glyceraldehyde-3-phosphate. In addition, the ischemic heart rapidly becomes acidotic (see later); this is due initially to the accumulation of lactate, which cannot be removed because of the decreased coronary flow, and to the release of protons when ATP is hydrolyzed.
Decreased ATP
ATP content falls rapidly after coronary occlusion, but does not reach levels low enough to deprive the substrate-binding sites of the contractile proteins and ion pumps of their supply of energy. This is because the normal cytosolic ATP concentration is 5 to 10 mM, whereas most substrate-binding sites are saturated at ATP concentrations less than 1 μM.
Attenuation of the allosteric effects of high ATP concentrations increases diastolic stiffness by inhibiting the dissociation of actin and myosin (see Chapter 4), and reduce contractility and impair relaxation by slowing ion pumps, ion exchangers, and passive ion fluxes through membrane channels (see Chapters 7 and 10). The sarcoplasmic reticulum calcium pump and plasma membrane sodium pump are also inhibited by a reduction in the allosteric effect of ATP (see Chapter 7). The former impairs relaxation, as does sodium pump inhibition, which causes a rise in cytosolic sodium that impairs relaxation by reducing calcium efflux via the Na/Ca exchanger. Sodium pump inhibition also reduces potassium influx and so decreases resting membrane potential; the latter, by inactivating sodium channel opening, slows conduction and provides a substrate for arrhythmias.
ATP depletion also decreases the free energy made available by hydrolysis of the terminal phosphate bond in ATP, which is proportional to the ATP/ADP ratio. Because even a slight fall in ATP concentration causes a disproportional increase in ADP concentration, energy starvation can reduce the free energy of ATP hydrolysis to levels that slow both the calcium pump of the sarcoplasmic reticulum and cross-bridge cycling (see Chapter 10).
Acidosis
Coronary occlusion causes a rapid fall in the pH within cardiac myocytes. Acidification is due largely to lactate formation and hydrolysis of ATP, which release weak acids (inorganic phosphate and lactate) that liberate hydrogen ions at the pH within the cytosol of cardiac myocytes. Some of these protons are absorbed when phosphocreatine hydrolysis releases creatine, which is a weak base, but the net effect of ischemia is rapid acidification of the heart.
Acidosis inhibits many important reactions in the heart; these include glycolysis (see Chapter 2), contractile protein interactions (see Chapter 4), most of the calcium fluxes that participate in excitation-contraction coupling and relaxation (see Chapter 7), and the cycling of plasma membrane ion channels responsible for cardiac action potentials (see Chapter 15). Another reason that acidosis causes arrhythmias is that protons increase internal electrical resistance by closing gap junctions in the intercalated disc, which slows conduction (see Chapter 13). Protons also interfere with many of the reactions involved in contraction and relaxation by competing with calcium for binding sites on troponin, ion channels, pumps, and exchangers (see Chapters 4 and 7).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


