Chapter 4 The Inflammatory Response
The Danger Hypothesis: Danger-Associated Molecular Patterns, Pathogen-Associated Molecular Patterns, and Alarmins
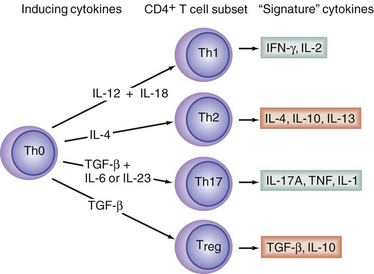
T helper cells (Th), which express the surface protein, CD4, also play key roles in the orchestration of innate and adaptive immune responses. Naïve CD4+ T cells (Th0 cells) can differentiate into at least four different Th subsets, called Th1, Th2, Th17, and T regulatory cells (Treg cells; Fig. 4-1). Th1 cells are responsible for directing the cell-mediated immune responses necessary for the eradication of intracellular pathogens, and favor macrophage activation. Th2 cells have been implicated in the pathogenesis of atopy and allergic inflammation and favor B cell growth and differentiation. Th1 cells produce the potent proinflammatory cytokines, interferon-γ (IFN-γ) and tumor necrosis factor-β (TNF-β; also called lymphotoxin). Th2 cells produce the cytokines interleukin-4 (IL-4), IL-5, IL-6, IL-10, and IL-13. The actions of IL-4, IL-10, and IL-13 are largely anti-inflammatory in nature. The actions of IL-6 can be both pro- and anti-inflammatory. Th17 cells produce several cytokines, notably IL-17A and IL-17F. Both IL-17A and IL-17F tend to be proinflammatory. The signature cytokines produced by Treg cells—namely transforming growth factor-β (TGF-β) and IL-10—are both anti-inflammatory. Thus, Th1 and Th17 lymphocytes are often viewed as being proinflammatory, whereas Th2 lymphocytes and Tregs are thought of as being anti-inflammatory. The cytokine, IL-12, drives Th1 differentiation, IL-4 induces Th2 differentiation, and TGF-β in combination with IL-6 promotes Th17 differentiation, but TGF-β in the absence of IL-6 promotes precursor cells to differentiate into Treg cells.1
Historically, activation of the immune system was thought to be triggered by the presence of antigens, which were recognized as being non-self in nature. However, the self-nonself model of immune surveillance and discrimination was burdened by the inability to account for numerous observations satisfactorily, such as the necessity for the presence of a tissue-damaging adjuvant to obtain a vigorous immune response to the nonself proteins present in vaccines. To address these concerns, the innovative immunologist, Polly Matzinger, formulated the danger model to explain immune system activation and discrimination.2 According to this hypothesis, which is now widely accepted, activation of the innate immune system is triggered by a diverse set of molecules that indicate the presence of danger to the host (i.e., something that could threaten health and well-being). Danger might come in the form of an invasion of host tissues by a pathogenic microorganism, but danger also might come in the form of trauma or malignant transformation. The molecules that signal the presence of something dangerous share a number of recognizable biochemical features, and collectively are referred to as danger (or damage)-associated molecular patterns (DAMPs). Some DAMPs are host-derived; compounds in this class are called alarmins.3 Other DAMPs are derived from pathogenic microorganisms and are called pathogen-associated molecular patterns (PAMPs).
Lipopolysaccharide
LPS is a complex glycolipid composed of a polysaccharide tail attached to a lipophilic domain called lipid A. The polysaccharide portion of the molecule tends to be structurally different in different species and strains of gram-negative bacteria, whereas the structure of lipid A (as well as a few neighboring sugar residues) is highly conserved across different species and strains of gram-negative microorganisms. A complex of LPS and a serum protein, LPS-binding protein (LBP), initiates the activation of monocytes and macrophages by binding to a surface protein, CD14. Because it is a glycophosphatidylinositol-anchored membrane protein, CD14 lacks a cytosolic domain and is unable to initiate intracellular signaling directly. Accordingly, investigators sought to identify another protein that presumably participates with CD14 to initiate the cellular response to LPS. The putative LPS coreceptor was ultimately identified as a Toll-like receptor (TLR).4
Toll-Like Receptors
In addition to LPS, other PAMPs and alarmins are recognized by various TLRs (Table 4-1). For example, TLR2 recognizes various bacterial lipoproteins, as well as peptidoglycan derived from gram-positive bacteria. TLR5 recognizes flagellin, a 55-kDa protein found in the flagella of certain bacteria. TLR9 recognizes certain oligonucleotides containing unmethylated CpG motifs that are more common in bacterial DNA than in mammalian DNA.
Table 4-1 Recognition of Pathogen-Associated Molecular Patterns and Alarmins by Pattern Recognition Receptors
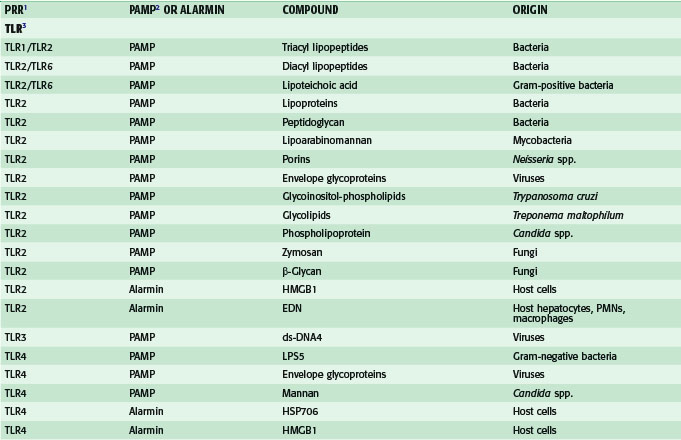
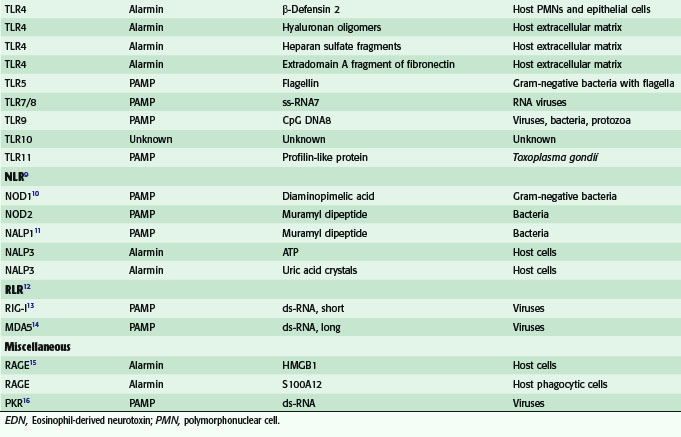
Among the TLRs, TLR4 seems to be particularly important, because this receptor recognizes not only the PAMP, LPS, but several endogenous danger signals as well. These endogenous ligands for TLR4 include the following: heat shock protein (HSP) 70, an inducible cytosolic protein, which is important for the proper folding of nascent proteins; high-mobility group box-1(HMGB1), an abundant DNA-binding protein, which is important for transcription and repair of DNA; extra domain A of fibronectin, an abundant protein in the extracellular matrix; and fragments of hyaluronan, a glycosaminoglycan, which is one of the chief components of the extracellular matrix. Some of these alarmins, such as HMGB1, are actively secreted by immunostimulated macrophages or enterocytes, whereas others, such as hyaluronan fragments, are probably generated as a consequence of trauma to tissues. Accumulating evidence obtained by the Billiar group at the University of Pittsburgh has suggested that many of the deleterious host responses to severe trauma and/or hemorrhagic shock are mediated by the interaction of endogenous alarmins with TLR4.4
Other Families of Pattern Recognition Receptors
In addition to members of the TLR family, there are two other families of PRRs that are important for recognizing DAMPs and initiating innate immune responses. These two families are the retinoid acid-inducible gene I (RIG-I)–like receptors (RLRs) and the nucleotide-binding oligomerization domain (NOD)–like receptors (NLRs).5 The two RIG-I–like receptors, RIG-I and melanoma differentiation-associated gene (MDA) 5, play a pivotal role in sensing the presence of viral double-stranded (ds) RNA in the cytoplasm. The interaction of ds-RNA with the C-terminal domains of RLRs initiates a signaling cascade, leading ultimately to the expression of cytokines important in antiviral immunity.
The two most extensively studied members of the NLR family of receptors are NOD1 and NOD2.5 These PRRs sense PAMPs derived from the synthesis and degradation of bacterial peptidoglycan. NOD1 is activated by diaminopimelic acid produced by gram-negative bacteria, whereas NOD2 is activated by muramyl dipeptide (MDP), produced by gram-negative and gram-positive bacteria. As will be discussed in greater detail, NLRs are not only important for sensing certain intracellular pathogens, but these receptors also play a key role in the processing for secretion of two important proinflammatory cytokines, IL-1β and IL-18.
The receptor for advanced glycation end products (RAGE) is a receptor that has multiple potential ligands, including HMGB1, amyloid-β peptide, and certain members of the S100-calgranulin family of proteins.6 Because RAGE-dependent signaling may be important for transducing some of the proinflammatory effects the alarmin, HMGB1, RAGE can be considered a PRR involved in innate immunity.
High-Mobility Group Box 1
When mice are injected with a lethal bolus dose of LPS, circulating levels of TNF peak approximately 60 to 90 minutes later and are almost undetectable within 4 hours. Although mice show clinical signs of endotoxemia (e.g., decreased activity and ruffled fur) within a few hours after the injection of LPS, mortality typically does not occur until more than 24 hours later, long after circulating levels of the so-called alarm phase cytokines, TNF and IL-1β, have returned to normal. These observations suggested the possibility to Wang and colleagues that LPS-induced lethality might be mediated by a previously unidentified factor that is released much later than TNF or IL-1β.6a Prompted by this idea, these investigators carried out a prolonged search for the putative late-acting mediator. This research program ultimately resulted in the identification of HMGB1 (formerly called HMG-1) as a novel mediator of LPS-induced lethality.
HMGB1 was originally identified in 1973 as a nonhistone nuclear protein with high electrophoretic mobility. A characteristic feature of the protein is the presence of two folded DNA-binding motifs termed the A domain and the B domain. Both these domains contain a characteristic grouping of aromatic and basic amino acids within a block of 75 residues termed the HMG box. HMGB1 has several functions within the nucleus, including facilitation of DNA repair and support of the transcriptional regulation of genes. When released by cells into the extracellular milieu, HMGB1 can interact with several different receptors, including TLR2, TLR4, and RAGE, on macrophages, endothelial cells, and enterocytes.7 Activation of these receptors leads to the release of other proinflammatory mediators, such as TNF and NO·.
Delayed passive immunization of mice with antibodies against HMGB1 confers significant protection against LPS-induced mortality. Furthermore, the administration of highly purified recombinant HMGB1 to mice is lethal. Thus, HMGB1 fulfills a modified version of Koch’s criteria for being a mediator of LPS-induced lethality in mice. Direct application of HMGB1 into the airways of mice initiates an acute inflammatory response and lung injury that is reminiscent of ARDS in humans. In addition, HMGB1 (or a truncated form of the protein, including only the B box domain) increases the permeability of human enterocyte-like monolayers in culture and promotes intestinal barrier dysfunction when injected into mice.8 Thus, it seems plausible that HMGB1 contributes to the development of organ dysfunction in human sepsis, a notion that is supported by the observation that circulating HMGB1 concentrations are significantly higher in patients with ultimately fatal sepsis than in patients with a less severe form of the syndrome.9 Circulating levels of HMGB1 are also increased in victims of trauma10 or burn injury.11 Administration of a neutralizing anti-HMGB1 antibody improves survival in mice subjected to lethal hemorrhagic shock.12 Ethyl pyruvate, a compound that blocks the release of HMGB1 from LPS-stimulated murine macrophage-like cells and inhibits release of the mediator in vivo, improves survival in mice with bacterial peritonitis, even when treatment with the compound is delayed for 24 hours after the onset of infection.13
Cytokines and Chemokines
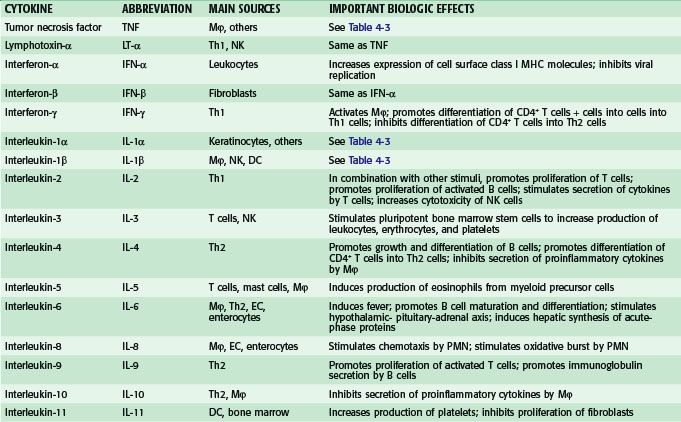
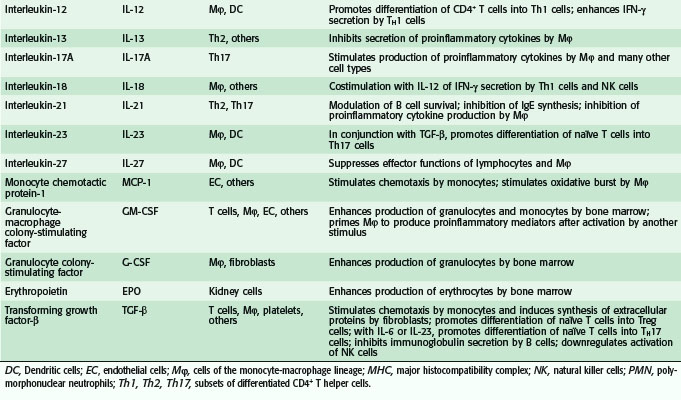
Overall, hundreds of soluble proteins involved in cell to cell signaling, variously called cytokines, chemokines, interleukins, colony-stimulating factors, and growth factors, have been identified and characterized. Some pertinent facts about some of the most important cytokines are provided in Table 4-2 and some of these mediators are discussed in greater detail in the sections that follow.
Interferon-γ and Granulocyte-Macrophage Colony-Stimulating Factor
When responsive target cells are exposed to IFN-γ, a number of genes are activated within minutes and without the synthesis of new copies of intermediate signaling proteins. IFN-γ–induced signal transduction occurs through the activation of a protein tyrosine phosphorylation cascade known as the JAK-STAT pathway (Fig. 4-2). JAK initially stood for “just another kinase” because the biologic role of these proteins was not established when they were initially discovered. Because these receptor-associated kinases look both outside and inside the cell, JAK has now come to stand for Janus kinases, after the two-faced Roman god. The moniker STAT, an acronym for signal transducers and activators of transcription, was appropriately chosen because, in medical parlance, an action to be carried out immediately is a stat order and signaling involving these proteins similarly occurs without delay. In addition to IFN-γ, a large number of other cytokines, including IL-6 and IL-11 (see later), also use versions of the JAK-STAT signaling mechanism. In mammals, there are seven mammalian STAT proteins (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) and four JAK proteins (JAK1, JAK2, JAK3, and TYK2).
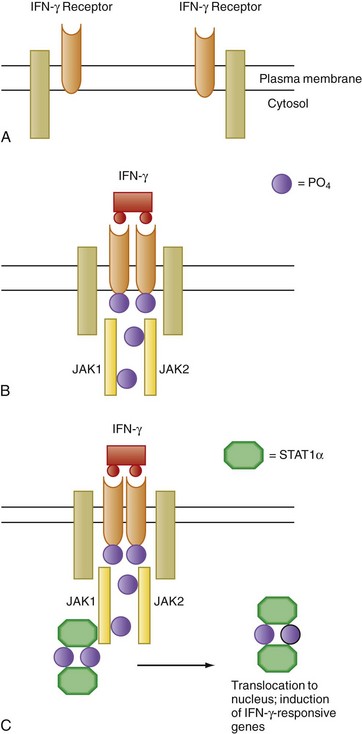
IFN-γR is a heterodimer that consists of a 90-kDa glycoprotein, the α chain, which is required for binding of the ligand, and a transmembrane protein, the β chain, which is required for signaling. Associated with the receptor are two members of the JAK family of kinases, JAK1 and JAK2. Interaction of IFN-γ with its receptor results in the dimerization of IFN-γR, which brings JAK1 and JAK2 into close association and leads to mutual phosphorylation and activation (see Fig. 4-2). The activated JAK kinases then catalyze the phosphorylation of tyrosine residues on the α chains of IFN-γR, which results in docking to the receptor complex by the transcription factor STAT1. After tyrosine phosphorylation, two copies of STAT1 form a homodimer (IFN-γ activation factor [GAF]) that subsequently dissociates from the receptor complex and translocates to the nucleus, where binding to the regulatory regions of target genes containing the IFN-γ activation site (GAS) nucleotide sequence leads to transcriptional activation.
JAK-STAT–dependent signaling is regulated in cells by a variety of mechanisms. Because STATs are activated by tyrosine phosphorylation, phosphotyrosine phosphatases are implicated in the negative regulation of JAK-STAT signaling pathways. In this regard, the first to be described were Src homology 2 domain (SH2)–containing tyrosine phosphatases such as SHP1 and SHP2. The presence of a characteristic amino acid sequence, the SH2 domain, in these cytoplasmic enzymes promotes the association of these phosphatases with phosphotyrosines present on activated receptors or on signaling molecules, as well as on activated JAKs.14 The transmembrane tyrosine phosphatase CD45, which is expressed on T and B cells, also downregulates JAK-STAT signaling. Two other important classes of proteins that regulate JAK-STAT signaling are the protein inhibitors of activated STAT (PIAS) and the inducible suppressors of cytokine signaling (SOCS).
A randomized trial of adjuvant treatment with recombinant GM-CSF in neonates with sepsis and neutropenia has shown that survival is significantly improved in the group treated with the cytokine–growth factor.15 Similarly, in a single-center randomized controlled trial (RCT), adjuvant treatment with recombinant GM-CSF significantly shortened hospital stay and decreased the number of infectious complications in patients with intra-abdominal sepsis.16 A more recent multicentric RCT has suggested that adjuvant treatment with GM-CSF can improve outcome for selected patients with sepsis.17 This study randomized 38 patients with severe sepsis and evidence of sepsis-induced immunosuppression to treatment with GM-CSF or placebo for 8 days. Although survival was similar in both groups, the GM-CSF–treated patients required mechanical ventilation and care in an ICU for a significantly shorter period of time
Crohn’s disease is a chronic inflammatory disorder of the gastrointestinal (GI) tract. Treatment with corticosteroids often ameliorates symptoms of the disease, but chronic administration of corticosteroids is associated with many adverse side effects. Accordingly, clinicians and scientists are actively seeking better approaches to treat Crohn’s disease. Because there is considerable evidence that Crohn’s disease may result, at least in part, from impaired innate immunity (e.g., caused by a mutation in the NOD2 gene),18 recombinant GM-CSF might be a therapeutic option for this condition. This hypothesis has been supported by the results from two RCTs, which showed that therapy with GM-CSF can induce remission in the absence of treatment with corticosteroids.19,20
Interleukin-1 and Tumor Necrosis Factor
IL-1 and TNF are structurally dissimilar pluripotent cytokines. Although these compounds bind to different cellular receptors, their multiple biologic activities overlap considerably. For example, in vitro, both cytokines are capable of activating endothelial cells, leading to increased expression of cell surface adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which play important roles in the process whereby neutrophils extravasate from circulation into tissues at the site of infection and/or inflammation. Similarly, incubating cultured monocytes, neutrophils, endothelial cells, hepatocytes, mesangial cells, articular chondrocytes, or synovial fibroblasts with IL-1 or TNF leads to secretion of a chemokine, IL-8 (see later), which is important for recruiting neutrophils into inflammatory foci. Recombinant forms of IL-1β and TNF have been available for many years. Table 4-3 summarizes some of the biologic effects, which are observed when human subjects are injected with recombinant IL-1β or TNF. The information in this table should convince the reader that many of the features associated with the systemic inflammatory response syndrome (SIRS), such as increased circulating leukocyte count and fever, can be reproduced by injecting subjects with the alarm phase cytokines, IL-1β or TNF. Through their ability to potentiate the activation of helper T cells, IL-1 and TNF can promote almost all types of humoral and cellular immune responses. Furthermore, both these cytokines are capable of activating neutrophils and macrophages and inducing the expression of many other cytokines and inflammatory mediators. Many of the biologic effects of IL-1 or TNF are greatly potentiated by the presence of the other cytokine.
Table 4-3 Partial List of Physiologic Effects Induced by Infusing Interleukin-1 or Tumor Necrosis Factor Into Human Subjects
| EFFECT | IL-1 | TNF |
|---|---|---|
| Fever | + | + |
| Headache | + | + |
| Anorexia | + | + |
| Increased plasma adrenocorticotropic hormone level | + | + |
| Hypercortisolemia | + | + |
| Increased plasma nitrite-nitrate levels | + | + |
| Systemic arterial hypotension | + | + |
| Neutrophilia | + | + |
| Transient neutropenia | + | + |
| Increased plasma acute-phase protein levels | + | + |
| Hypoferremia | + | + |
| Hypozincemia | + | |
| Increased plasma level of IL-1RA | + | + |
| Increased plasma level of TNF-R1and TNF-R2 | + | + |
| Increased plasma level of IL-6 | + | + |
| Increased plasma level of IL-8 | + | + |
| Activation of coagulation cascades | − | + |
| Increased platelet count | + | − |
| Pulmonary edema | − | + |
| Hepatocellular injury | − | + |
Interleukin-1 and the Interleukin-1 Receptor
Transgenic mice deficient in ICE–caspase-1 are resistant to endotoxic shock and manifest an impaired ability to mount a local inflammatory response to intraperitoneal zymosan, a known inducer of sterile peritonitis. In contrast, ICE–caspase-1 knockout mice manifest increased susceptibility to infections caused by various pathogens, including E. coli, Shigella flexneri, Salmonella typhimurium, Listeria monocytogenes, and Candida albicans. Taken together, these data suggest that ICE-dependent processes, including secretion of the mature forms of IL-1β and the related cytokine, IL-18 (see later), are important for host defense against microbial infection but also are crucial for the pathologic manifestations of poorly controlled inflammation.21 Various ICE-like enzymes, the caspases, have been identified as being important mediators of the process of programmed cell death, or apoptosis. A special form of apoptosis, called pyroptosis, can occur within minutes after macrophages are infected with certain intracellular pathogens. Pyroptosis is an ICE-dependent process.
The activation of ICE–caspase-1 can be triggered in cells by the formation of a molecular complex called the inflammasome.21 Inflammasomes are oligomeric complexes, which are composed of ICE–caspase-1 as well as various members of the NLR family of PRRs called NALPs (NACHT domain leucine-rich repeat and PYD-containing protein) and an adapter protein called ASC (apoptosis-associated specklike protein containing a CARD). Assembly of the inflammasome, which in many cases is triggered when an NLR family member senses the presence of PAMP molecules and ultimately leads to ICE–caspase-1 activation and secretion of IL-1β (and IL-18). Inflammasomes that contain a particular NALP (NALP3), can activate ICE–caspase-1 in response to a wide variety of unrelated compounds, including certain toxins, high concentrations of adenosine triphosphate (ATP), and crystals of monosodium urate (the mineral-like structures that are associated with gout). Alum, the adjuvant used in most vaccines to enhance immune responses to antigens, also has been shown to induce activation of the NALP3 inflammasome. All these compounds can lead to ICE–caspase-1 activation and secretion of IL-1β and the related cytokines, IL-18 and IL-33.
Because the cytoplasmic TIR domains of the TLRs are homologous to the cytoplasmic region of IL-1RI, it is not surprising that some shared mechanisms are responsible for downstream signaling (Figs. 4-3 and 4-4). In the MyD88-dependent pathway, an adapter protein, myeloid differentiation primary response factor 88 (MyD88), links the receptor to another protein called IL-1 receptor–associated kinase 1 (IRAK-1). On binding of the ligand to the TLR (or IL-1RI), IRAK-1 is phosphorylated and dissociates from the receptor complex, thereby allowing it to interact with another signaling protein, TNF receptor–activated factor 6 (TRAF6). This process results in the activation of nuclear factor κB (NF-κB), a pivotal proinflammatory transcription factor, as well as the phosphorylation signaling cascades involving mitogen-activated protein kinases (MAPKs).
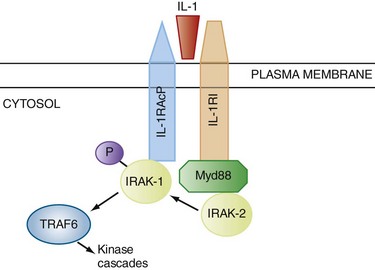
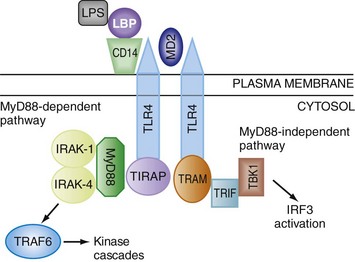
FIGURE 4-4 Simplified representation of the intracellular signal transduction steps, which are initiated by the binding of the microbial product, LPS, to TLR4. The interaction of LPS with TLR4 requires several extracellular accessory proteins—LBP, CD14 (a glycophosphoinositol-anchored cell surface receptor), and MD2. After assembly of the extracellular LPS-LBP-CD14-TLR4-MD2 complex, signaling can follow two different pathways. In the more immediate MyD88-dependent signaling pathway, an adapter protein, MyD88, links the intracellular portion of TLR4 to other adaptor proteins called IRAK-1 and IRAK-4. Phosphorylation of IRAK-1 allows it to dissociate from the receptor complex, thereby permitting it to interact with another signaling protein, TRAF6. This process results in the activation of NF-κB, a pivotal proinflammatory transcription factor, as well as signaling cascades involving MAPKs.6 In the more delayed MyD88-independent pathway, the adapter proteins, TRIF and TRAM, lead to the activation of the serine-threonine kinase, TANK-binding kinase (TBK) 1, which leads to the activation of the transcription factor, IRF3. After phosphorylation, IRF3 forms a complex with cyclic adenosine monophosphate (cAMP) response element-binding (CREB) protein-binding protein (CREBBP), and this complex translocates to the nucleus, leading to the transcription of the genes for IFN-α and IFN-β, as well as other interferon-induced genes. The association of TRIF with the TIR domain of TLR4 also leads to the activation of NF-κB via pathways, which involve TRAF6 as well as another adapter protein called RIP1 (not shown).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


