The Heart in Muscular Dystrophies
Philip T. Thrush
Jerry R. Mendell
Kevin M. Flanigan
Timothy M. Hoffman
Hugh D. Allen
Muscular dystrophy (MD) may be associated with cardiomyopathy (CM) (sometimes dilated), conduction disturbances, or both. Depending on the type of dystrophy, and possibly the location of the gene abnormality, expression can be variable. With the advent of genetic testing, these diseases are being studied much more systematically and vigorously. The result is that more specific information about each type and subtype has been gained. These newer understandings about the diseases and their nuances will ultimately lead to improved diagnosis and likely different classifications and therapeutic approaches.
Muscular Dystrophies with Clinical Cardiac Disease
Myotonic MD is the most common form of MD (inclusive of both adults and children). On the basis of new molecular insights, at least two variants must be considered (1,2). The classic disorder is referred to as dystrophic myotonia type 1 (DM1). It is associated with a CTG repeat in the 3′ untranslated region of dystrophia myotonica–protein kinase gene. Repeat size instability leads to anticipation in disease onset from one generation to the next, and the children of mothers with DM1 can have congenital dystrophia myotonia, a severe grave form of the disease in which the CTG repeat is far more expanded. In these infants, the cardiac manifestations are considerably more severe (dilated cardiomyopathy [DCM], cardiac rhythm disturbances, and sudden death). A second variant of myotonic dystrophy is referred to as DM2. Several notable differences from the classic disorder start with the molecular defect characterized by a CCTG expansion in intron 1 of the zinc finger protein 9 (ZNF9) gene. The cardiac manifestations also include conduction system disease as in DM1. The congenital form of the disease is not seen in DM2.
Dystrophin deficiency causes the second largest group of dystrophies with cardiac involvement (and the most common in childhood). The severity of the skeletal muscle disease correlates with the quality and quantity of dystrophin, the product of the X-linked DMD gene. Duchenne muscular dystrophy (DMD) is the most severe variant, and is typically associated with <5% of normal levels of dystrophin in skeletal muscle. Becker muscular dystrophy (BMD) is a milder form of dystrophinopathy, in which greater amounts of dystrophin protein are present in the muscle. These differences in dystrophin expression are due to differing DMD mutations that either truncate protein expression or allow expression of protein of diminished size or amount.
Another unique disorder with prominent cardiac manifestations is Emery–Dreifuss muscular dystrophy (EDMD). EDMD refers to a syndrome caused by more than one gene defect. The classic disease, X-linked EDMD (XL-EDMD), is marked by striking contractures (particularly at the elbow) and in most cases is caused by a mutation of the STA gene that encodes emerin, a protein of the inner nuclear membrane. Recently, another X-linked gene FHL1 has been demonstrated to be the cause of about 10% of XL-EDMD (3). Mutations in the LMNA gene are responsible for autosomal forms of EDMD, which occur most commonly as autosomal dominant (AD) and occasionally as autosomal recessive (AR) traits. Both are almost identical in signs and symptoms to the X-linked form. The LMNA gene encodes alternatively spliced isoforms of type A lamins, lamin A and lamin C (4). Lamins are found on the inner nuclear membrane where they interact directly with other proteins including emerin, seeming to account for the overlapping clinical picture. Perhaps adding to the confusion is a DCM occurring in the absence of MD also associated with LMNA mutations. On the other end of the spectrum, adding further complexity to the LMNA phenotypes, is a generalized form of weakness affecting pelvic and shoulder muscles referred to as limb-girdle muscular dystrophy 1B (LGMD1B). It is clinically distinct from EDMD because of lack of extremity contractures. Of particular relevance, LMNA mutations causing LGMD1B, EDMD, and DCM all carry a risk of atrial arrhythmia, CM with heart failure, and sudden death (probably owing to tachyarrhythmias) (5).
The LGMDs are a clinically and genetically heterogeneous group of disorders. LGMDs are classified based on the mode of inheritance and include both AD (LGMD1) and AR (LGMD2) forms. Based on linkage analysis, letters of the alphabet have been sequentially assigned to these disorders depending on the order in which linkage was established. Thus far, eight types of LGMD1A to 1H and 23 subtypes of LGMD2A to 2W have been identified and classified (6,7). Patients usually have pelvic girdle weakness with varying degrees of shoulder girdle muscle weakness. Mild contractures are seen with LGMDs but are never the predominant clinical feature as seen in EDMDs. The LGMD phenotypes with CM are described later.
A comment about the term X-linked cardiomyopathies is necessary here for clarification. This is a clinical term that did have value in the premolecular genetic era, but with our current degree of understanding regarding specific gene defects, it is best to consider the major X-linked cardiomyopathies separately. Four separate genetic disorders with cardiac and skeletal myopathies are found on the X chromosome. DMD and BMD already have been discussed. These disorders, caused by the same gene mutation with varying degrees of dystrophin deficiency, are linked to the short arm of the X chromosome at Xp21. The other major CM on the X chromosome, EDMD (8), with a mutation in the STA gene, is linked to the long arm of the X chromosome at Xq28. A third CM on the X chromosome is Danon disease (9), also known as X-linked vacuolar CM and myopathy, which is caused by a mutation in the gene encoding lysosome-associated membrane protein-2 (LAMP2) at Xq24. This form of dystrophy can have hypertrophic CM. The final disorder of this group is Barth syndrome (10). It is a mitochondrial disease caused by a mutation of the tafazzin gene associated with decreased amounts and altered structure of cardiolipin, the main phospholipid of the inner mitochondrial membrane. The gene maps to the long arm of the X chromosome at Xq28. Patients have variable clinical findings, often including heart failure, CM, neutropenia, and growth retardation.
Assessment Tools
Electrocardiography
Holter Monitoring
Twenty-four hour electrocardiography gives insight into arrhythmias and disorders of conduction seen in patients who may be asymptomatic. It also allows measurement of heart rate variability in patients with disordered automaticity.
Echo Doppler Studies
Somatic deformities associated with various forms of dystrophy may include chest wall deformities and kyphoscoliosis that distort the relationship of lungs and heart, creating challenges for ultrasound evaluation. It is important to record and measure the left ventricular dimensions in diastole and systole, areas of dyskinesis and akinesis, anterior mitral valve leaflet E-point septal separation (EPSS), shortening fraction (SF), ejection fraction (EF), and rates of wall contraction and relaxation. Planimiterization of two-dimensional images allows fairly accurate predictions of left ventricular volumes and EF. The sphericity index, derived from comparing the long-axis left ventricular dimensions in diastole and systole with the chord from the mitral annulus to the apex in diastole and systole, has been used to quantify the myopathy. The value should be <0.66. If it approaches 1.0, it indicates that the chamber is rounded instead of elliptical (remodeled), and a dilated myopathy is likely (11). Newer echocardiographic modalities, such as strain and strain rate, may identify early manifestations of CM, but the time-intensive off-line processing of these images remains a limitation to their routine use in the clinical setting (12,13,14).
Friedberg and Silverman have defined criteria for DCM that include prolonged systolic duration and shortened diastolic duration (15). Doppler mitral and tricuspid regurgitation jet durations were used to measure systole. Diastole was measured as the time from cessation of the regurgitation jet to the onset of the next atrioventricular (AV) valve regurgitant jet. A ratio of systolic time/diastolic time was derived and corrected in relation to heart rate. The tricuspid systolic interval/RR interval in normal subjects was 0.41 ± 0.07 (mitral, 0.42 ± 0.08), and the systolic/diastolic ratio in normal subjects was 0.77 ± 0.24. There are few studies in MD that have used this method, but it holds potential as another useful measurement. See Chapter 9 for a detailed description of functional testing by echo and Doppler techniques.
Doppler analysis is used to demonstrate mitral and aortic valve regurgitation. Quantification of tricuspid regurgitation allows prediction of systolic right ventricle and pulmonary artery pressures. Evaluation of pulmonary regurgitation allows estimation of pulmonary arterial diastolic pressures. It is also possible to estimate cardiac output by planimiterization of the pulsed Doppler aortic waveforms with respect to space and time. Newer studies, especially in adult patients, and a few in the pediatric population, have used Doppler tissue analysis to evaluate wall contractile function and diastolic function. These approaches are beginning to have usefulness for evaluation of CM in the MD population.
Because many of these patients cannot exercise physically, dobutamine stress echo studies may provide information regarding myocardial performance, especially in patients with borderline normal resting studies. These studies should be performed carefully, recognizing that atrial and ventricular tachyarrhythmias could occur during such stress testing.
Other Imaging Methods
Other tools can be used to measure left ventricular volumes and derivation of EFs when patients cannot have adequate echo studies; these include magnetic resonance imaging (MRI), computerized tomography imaging, radionuclide imaging, and positron emission testing. Each has advantages and disadvantages, including cost, accessibility, and limitations, for example, the presence of metal rods in patients with previous scoliosis surgery may interfere with MRI analysis. There may be variability in the information that can be derived from these other imaging methods depending on the availability of the most recent equipment and the sophistication of the software used for analysis. However, the use of these modalities may provide additional information that cannot be garnered from echocardiography, such as the identification of myocardial fibrosis using late-gadolinium enhancement with cardiac MRI.
Muscular Dystrophies with Cardiac Involvement
Dystrophinopathies
DMD and BMD are related disorders (16,17,18), differing in severity because of the amount or quality of the expressed dystrophin protein. In nearly all cases, DMD patients express <5% dystrophin in skeletal muscle biopsies, an amount insufficient to maintain ambulation much beyond age 12. In contrast, in BMD, the gene mutation permits varying degrees of dystrophin expression that can be quantified on skeletal muscle biopsy. In most cases, both the quantity and the size of dystrophin are reduced. The amount of dystrophin expressed also can be determined by cardiac biopsy, but this is not a common or practical approach. It is important to understand that BMD covers a very broad spectrum of disability from mild to severe depending on the specific gene mutation. It is also important to note that skeletal and cardiac muscles may exhibit paradoxical clinical responses. In DMD with severe skeletal muscle loss early in the course of the disease related to almost complete dystrophin deficiency, patients lose ambulation between 10 and 12 years of age and have a sedentary wheelchair-dependent lifestyle. In this environment, there may be very little stress on the heart on a day-to-day basis. This probably accounts for the uncommon presence of symptoms and signs of clinical heart failure, which often do not manifest without a catastrophic event, such as a life-threatening pulmonary infection. In contrast, the patient with BMD who has CM will have more skeletal muscle and preserved ambulation that create more cardiac demands, leading to symptoms of heart failure that mandate treatment.
Dystrophinopathies: Duchenne Muscular Dystrophy
Boys with DMD have few problems in the neonatal period. Some have delayed onset in walking independently. They sometimes crawl later than average. The disease usually is not recognized until about 3 years of age because the affected boys run and jump poorly and cannot keep up with other children in normal play activity. However, DMD can be identified using newborn screening, although this is not a standard practice currently (19). If there is a known family history, for example, siblings with DMD, a new male infant may be recognized as having DMD with creatine kinase (CK) and genetic confirmation. By school age, the difference in muscular function becomes very apparent. Stair climbing usually requires a handrail, and they usually climb stairs one step at a time rather than alternating from step to step. Falling becomes an increasing problem. Facial injuries can result from forward falls because the arms are too weak to brace against the fall. To get up from the floor, boys roll to a prone position, spread their legs for balance, first lift their buttocks and then “walk” their hands up their legs (Gowers’ sign). By age 6 to 7 years, most boys assume a waddling gait with a lordotic posture. Walking becomes increasingly difficult about age 10, and without intervention, most of these patients will become wheelchair dependent by about age 12. For the DMD population at large, the IQ is reduced by approximately one standard deviation (SD) from the normal population. Cognitive difficulties affect verbal and memory skills selectively.
On physical examination, certain features are easily recognized and include the following: enlarged (hypertrophied, not “pseudohypertrophied”) calf muscles that feel rubbery on palpation; weak
neck muscles that cannot raise the head from supine, especially if the neck is first hyperextended; and weakness of proximal muscles of varying degrees, always worse than distal muscle. Late in the course, all muscle function is impaired except for minimal hand movement. Diaphragm and intercostal muscles are compromised, leading to impaired airway clearance due to poor cough, aspiration, and predisposition to pneumonia. A typical scenario is an episode of pneumonia that increases cardiac demands leading to heart failure. Some patients may have a tachyarrhythmia at any time prior to this. Death has historically occurred sometime in the early 20s, although aggressive and early use of nocturnal ventilatory support may have created a significant impact in extending life expectancy (20).
neck muscles that cannot raise the head from supine, especially if the neck is first hyperextended; and weakness of proximal muscles of varying degrees, always worse than distal muscle. Late in the course, all muscle function is impaired except for minimal hand movement. Diaphragm and intercostal muscles are compromised, leading to impaired airway clearance due to poor cough, aspiration, and predisposition to pneumonia. A typical scenario is an episode of pneumonia that increases cardiac demands leading to heart failure. Some patients may have a tachyarrhythmia at any time prior to this. Death has historically occurred sometime in the early 20s, although aggressive and early use of nocturnal ventilatory support may have created a significant impact in extending life expectancy (20).
Cardiac Pathology
Cardiac involvement is universal with an estimated incidence of CM as high as 25% by age 6 years and 59% by age 10 years (21,22). However, more recent studies suggest the median age of diagnosis of CM in these boys is 14 to 15 years (23,24). There is epimyocardial muscle replacement with fat and fibrosis beginning in the left ventricular posterior wall behind the posterior mitral annulus. Histologic studies show that the fibrosis begins at the epicardium and progresses toward the endocardium (25). The myocardial scarring progresses apically and ultimately invades the septum (26). The right ventricle and the atria are seldom involved. The fibrosis leads to dysfunction and occasional dilation, especially with severe dysfunction, with initial thinning and even outpouching of the left ventricular posterior wall behind the mitral area to a severe generalized CM. It is possible that the left ventricle cannot dilate if the wall is severely fibrotic, creating a restrictive myopathy. If dilation is present, the patient can develop mitral regurgitation and occasionally, aortic regurgitation. With left ventricular failure, there can be secondary pulmonary hypertension and right ventricular failure associated with pulmonary and tricuspid regurgitation. In addition, left ventricular noncompaction (LVNC), a form of CM in which there are deep trabeculations within the left ventricular myocardium, may be seen in a significant number of these patients (27,28) and may portend a poorer prognosis (28). Fibrosis and fatty infiltration can also involve the conduction system, including the sinoatrial (SA) node and AV node (29,30).
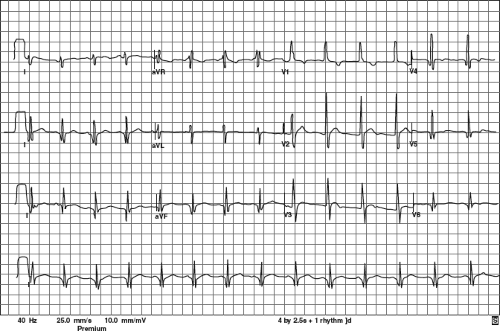 Figure 57.1 ECG from a 20-year-old patient with DMD. Note the shortened PR interval; Q-waves in II, III, aVF, V5, and V6; intraventricular conduction delay; and RVH. The QTc was 430 ms. |
Cardiac History and Physical Examination
The DMD patient has very few cardiac complaints, mainly because of physical inactivity. Even those with severe CM have few symptoms, other than shortness of breath, which can also be due to respiratory compromise from their weak chest and diaphragmatic musculature. Some will have paroxysmal nocturnal dyspnea. Others may detect palpitations if they have ventricular or atrial arrhythmias.
It is important to realize that patients with DMD and BMD can have severe complications from anesthesia, including cardiac arrest. Most complications seem to be related to use of succinylcholine, a muscular relaxant that may trigger hyperkalemia (31). Others have been attributed to use of volatile anesthetic agents. Patients can also have a reaction similar to malignant hyperthermia (31), develop rhabdomyolysis, and have masseter muscle spasm. It is apparent that anesthesia must be approached with caution in patients who have dystrophinopathies (31,32).
The cardiac examination is seldom abnormal, even in the presence of CM. Occasionally, third or fourth heart sounds may be present. According to Perloff et al. (33), a pulmonary outflow murmur is present in most patients, but that has not been our experience. Some will have a click and murmur of mitral valve prolapse; we have heard this in patients with severe chest wall deformities secondary to scoliosis. There may be neck vein distention, peripheral edema, or sacral edema. The overall examination is often distorted by chest wall deformities, especially in older patients who have scoliosis.
Electrocardiographic Features
Historically, the characteristic ECG in DMD is described as having a shortened PR interval, deep Q-waves in leads I, aVL, V5, and V6, and occasionally in leads II, III, and aVF. There is often a tall right precordial R-wave and an increased R/S ratio (30,34) (Fig. 57.1).
Some have reported QT prolongation (21,35) and QT dispersion abnormalities (36).
Some have reported QT prolongation (21,35) and QT dispersion abnormalities (36).
A recent prospective study of 115 boys with DMD (40 with CM) confirmed that the most commonly seen findings were (a) short PR interval (43%), (b) right ventricular hypertrophy (RVH) (37%), (c) prominent Q-waves in leads V5 (34%) and V6 (33%), (d) Q-waves in the inferior lateral leads in only 9, and (e) Q-waves in leads I, aVL, V5, and V6 in only 3. None had prolonged QTc intervals, two had ST-T depression (1 with CM) and 38 had flat/biphasic ST segments (15 with CM). The ECG did not discriminate the CM subgroup from the DMD boys who had EF >55% (37).
A recent serial analysis of 154 DMD boys (91 with CM) studied prospectively over a 9.4-year period with a total of 805 ECGs (367 in CM group) showed no correlation of RVH amplitude with echocardiographic LVID, EPSS, SF, or EF. Thus RVH does not correlate with CM (38).
Another recent retrospective study of 150 MD patients, including 86 DMD boys (51 with CM defined as EF <55% or LVIDd >2SD = 59%) showed that abnormalities (mainly repolarization artifacts, ST-T changes, RVH, or BVH) correlated well with a general MD population who had CM as previously defined. The DMD population was not separated in the retrospective paper’s data analysis but abnormalities were seen in only 63/213 (29.5%) of the DMD ECGs that were analyzed. There was no mention of shortened PR interval or correlation with CM in the DMD subpopulation (39).
Holter analysis has shown that automaticity is also affected whereby there is a resting sinus tachycardia, loss of circadian rhythm, and reduced heart rate variability in many patients with DMD (40,41,42,43,44). These findings are typically referred to as disordered automaticity. There are frequent arrhythmias in older patients including ectopic atrial tachycardia, atrial fibrillation, transient second- and third-degree AV block, and more ominous ventricular tachycardias (40). One publication showed that there was a predictive ECG pattern in terminal DMD consisting of RV1 <0.6 mV, RV5 <1.1 mV, and RV6 <1.0 mV; abnormal T-waves in II, III, aVF, V5, and V6; conduction disturbances; premature ventricular contractions (PVCs); and sinus tachycardia (45). The presence of multiform PVCs and ventricular tachycardia on Holter monitoring portends possible sudden death due to ventricular fibrillation (46,47,48).
Doppler Echocardiogram and Other Imaging Studies
The echocardiographic findings in boys with DMD correlate with the autopsy findings of posterior epicardial thinning leading to ultimate DCM. The first descriptions were by Goldberg et al. (48,49,50,51), who showed thinner left ventricular posterior walls, especially behind the posterior mitral valve leaflet, diastolic dysfunction, contraction abnormalities that progressed inferiorly, and temporally progressive wall thinning in these patients during serial studies. Other serial studies showed progressive deterioration toward left ventricular dilation and dysfunction by evaluation of left ventricular diameter changes (21) (Fig. 57.2), systolic time interval changes (49,50,51), and development of mitral valve prolapse (47).
Findings of CM do not necessarily parallel the progressive skeletal muscle changes (52,53). Increasing left ventricular diastolic and ultimately systolic volumes, decreasing shortening fraction and EFs, and increasing sphericity indices (11,54) (Fig. 57.3) indicate worsening CM (54,55,56). Tissue Doppler echocardiography studies have shown decreased systolic contraction and diastolic relaxation rates in patients with DMD, even when they had otherwise normal echocardiographic findings (55,56). Further studies using this technology are necessary to confirm these findings, but it appears to be promising for assessment of these populations.
One study using equilibrium radionuclide angiography in 9- to 18-year-old patients with DMD showed normal resting EFs (>50%) in 79% of the patients and normal right ventricular EFs (>45%) in 95%. However, when they used dobutamine perfusion, there were marked age-related decreases in both left and right ventricular EFs (57). Another 5-year follow-up serial radioisotope study showed resting changes in the septum that portended a fatal outcome (58).
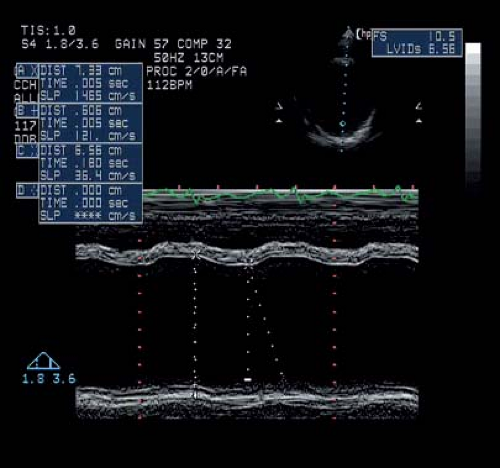 Figure 57.2 M-mode tracing from a 24-year-old patient with advanced DMD. His E-point septal separation was nearly 2 cm (normal is <5 mm). |
MRI and angiography hold great promise for detection of scarring and fibrosis within the muscle (59,60,61) (Fig. 57.4) and quantification of left ventricular function by using circumferential strain analysis (59,62,63,64) (see Chapter 10). The detection of myocardial fibrosis by late-gadolinium enhancement, also known as delayed myocardial enhancement, has been shown to identify patients at higher risk for ventricular arrhythmias, lower EF, adverse left ventricular remodeling, and death (65). The presence of non–steel-containing Harrington rods is not a contraindication for MRI evaluation. These
studies have shown that fibrosis and abnormal circumferential strain are noted long before EF abnormalities are seen on echo.
studies have shown that fibrosis and abnormal circumferential strain are noted long before EF abnormalities are seen on echo.
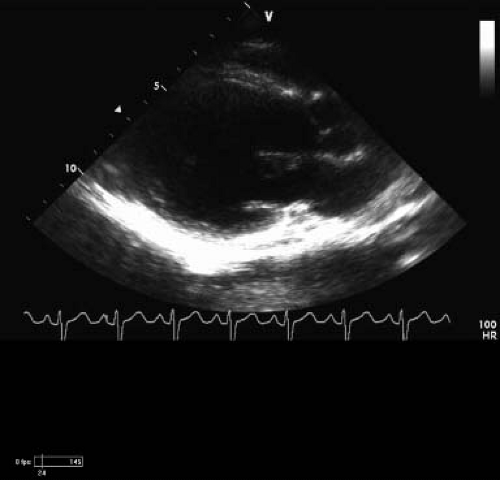 Figure 57.3 Two-dimensional long-axis echo from patient in Figure 60.2. Note the dilation of the left ventricle at 4.6 Z score (SDs). His EF on afterload reduction and β-blocker treatment is 39%, and SF is 19%. The sphericity index is 0.8 (normal is <0.66). He has posterior wall thinning, and that area is nearly akinetic. |
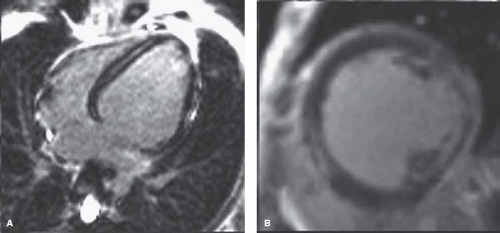 Figure 57.4 MRI from the same patient as in Figures 58.2 and 58.3. A: Note that fibrosis (white appearance) posterior to the mitral papillaries has nearly completely replaced muscle. B: Fibrosis extends through the myocardium. Functional information was similar to the echo results. (Courtesy of Stephen Cook, MD.) |
Neuroendocrine Abnormalities
Plasma α-atrial natriuretic peptide (α-ANP) and brain natriuretic peptide (BNP)/N-terminal proBNP (NT-proBNP) levels are elevated in adults with congestive heart failure. A few studies of patients with DMD have shown these to be elevated in patients with echocardiographic evidence of left ventricular dysfunction (EF ≤15%) or end-stage cardiorespiratory failure (66,67). However, a more recent study demonstrated no correlation between NT-proBNP levels and DCM in DMD and BMD patients or carriers (68). More studies are necessary to clarify the meaning of these findings, but ANP and BNP levels seem to be a promising method for evaluating these patients and assessing the patient’s response to treatment (see Chapter 73).
Therapy
The approaches to treatment include symptomatic, preventive, and curative. Most emphasis had been on treating symptoms, but they are seldom present in DMD boys. In the past, the empiric standard treatment was use of digitalis and diuretics. Digitalis has fallen out of favor with many cardiologists who treat DMD because of its proarrhythmic potential.
A recent consensus statement recommends doing echo examinations after the age of 6 years and repeating them at least every 2 years thereafter until 10 years of age, then annually. “Use of standard heart failure interventions with deterioration of function”—“even if asymptomatic”— was also recommended (69).
More recently, afterload reduction with angiotensin-converting enzyme inhibitors (ACE inhibitors) has been used with some success (53). Some have advocated using ACE inhibition for patients with EF <55%, left ventricular dilation (>2 Z scores/body surface area [BSA]), sphericity index (>0.66), and/or an abnormal tissue Doppler myocardial performance index (<0.35) (53,57,58,70). Jefferies et al. (53) reported that ACE inhibition (and sometimes additional β-blocker treatment if the echocardiographic indices of remodeling did not improve after 3 months) resulted in stabilization in 2 of 29 boys, improvement in 8 of 29, and normalization in 19 of 29 (16 Duchenne, 3 Becker). Others have evaluated a similar population with EF <55% who were treated with ACE inhibition with varied results (70). Some controversy exists as is evident from a letter to the editor responding to this study with many important questions about the true effect of the drug, selection bias, and variable expressions of the disease (71).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


