Rodney H. Falk, Ray E. Hershberger
The Dilated, Restrictive, and Infiltrative Cardiomyopathies
There is, at present, no universal definition of cardiomyopathy. Even though it is now agreed that myocardial disease secondary to atherosclerotic coronary artery disease, valvular disease, congenital heart disease, and systemic hypertension should not be classified as a cardiomyopathy, opinion differs whether the condition should be defined on the basis of morphology and whether molecular disturbances such as the channelopathies should be included. An American Heart Association definition1 describes cardiomyopathies as “a heterogenous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilation and are due to a variety of causes and frequently are genetic. Cardiomyopathies either are confined to the heart or are part of a generalized systemic disorder often leading to cardiovascular death or progressive heart failure-related disability.” This classification includes patients with predominantly electrical dysfunction of the heart, a group not included in a European Working Group definition.2 Both U.S. and European experts, however, have recognized the growing importance of genetics in patients with cardiomyopathy since these position papers were released.
The ability to combine genetic information with phenotypic information regarding left ventricular (LV) structure and function forms the basis of cardiovascular genetic medicine (Fig. 65-1). Molecular genetic testing in patients with cardiomyopathies not only enhances the care of symptomatic patients but will also benefit asymptomatic patients and family members through proper risk assessment. Moreover, it is likely that in the future, genomic information will predict the natural history, as well as guide therapy. However, the expansion of clinical genetic testing that has been made possible by next-generation sequencing also brings new challenges in terms of knowing which tests to order, how to conduct pretest counseling and obtain consent, and how to interpret molecular genetic test results. Table 65-13,4 presents an overview of the classification of cardiomyopathies based on phenotypic (the “phenome”) and genotypic information. The “phenome” includes data on cardiac morphology, physiology, and cellular and molecular pathology, as well as on other aspects of the environment relevant to the specific disease in question.5 Despite the integration of genetics and genomics, the phenotypic information derived from information about LV chamber size and function remains highly relevant in terms of clinical care despite the absence of universally accepted definitions. Although this chapter focuses primarily on cardiomyopathies that are not associated with other clinical syndromes (i.e., “nonsyndromic”), there are multiple syndromes in which a cardiomyopathy develops in concert with multiorgan system involvement. Hypertrophic cardiomyopathy (HCM; see Chapter 66) is also mentioned briefly herein because of its significant genetic overlap with dilated cardiomyopathy (DCM) and restrictive cardiomyopathy (RCM). It is also important to recognize that although myocardial dysfunction as a result of hypertension and ischemic heart disease must be differentiated from the cardiomyopathies, they often coexist and may aggravate an underlying primary cardiomyopathy.

TABLE 65-1
Classification of the Cardiomyopathies by Phenome and Genome
| TYPE | PHENOME | GENOME | ||||
| Morphology | Physiology | Pathology | Systemic Conditions or Diseases, Clinically Relevant Features, Classic Risk Factors, Associations | Nonsyndromic, Usually Single Gene | Syndromic | |
| Dilated (DCM) | LV/RV dilation with minimal or no wall thickening | Reduced contractility is the primary defect; variable degree of diastolic dysfunction | Myocyte hypertrophy; scattered fibrosis | Hypertension; alcohol use; thyrotoxicosis, myxedema; persistent tachycardia; toxins, e.g., chemotherapy, especially anthracyclines; radiation; pregnancy | Diverse gene ontology (see Table 65-2) with >30 genes implicated (see also Table e65-1 ) | Diverse array of associated conditions, especially muscular dystrophies (MDs): Emery-Dreifuss MD, limb-girdle MD, Duchenne/Becker MD; Laing distal myopathy; Barth syndrome; Kearns-Sayre; others3,4 |
| Restrictive (RCM) | Usually normal chamber sizes; minimal wall thickening | Contractility normal or near-normal with a marked increase in end-diastolic filling pressure | Specific to type, diagnosis: amyloid, iron, glycogen storage disease, others | Endomyocardial fibrosis, amyloid, sarcoid, scleroderma, Churg-Strauss syndrome, cystinosis, lymphoma, pseudoxanthoma elasticum, hypereosinophilic syndrome, carcinoid | If not associated with a systemic genetic disease (e.g., hemochromatosis), genetic cause found most commonly to result from sarcomeric gene mutations (see Table e65-1 ) | Gaucher disease, hemochromatosis, Fabry disease, familial amyloidosis. Mucopolysaccharidoses, Noonan syndrome |
| Hypertrophic (HCM) | Usually normal or reduced internal chamber dimension; wall thickening pronounced, especially septal hypertrophy | Systolic function increased or normal | Myocyte hypertrophy, classically with disarray | Severe hypertension can confound clinical, morphologic diagnosis | Mutations of genes encoding sarcomeric proteins (Chapter 66; also see Table e65-1 ) | Noonan/Leopard, Danon, Fabry, WPW, Friedrich ataxia, MERRF, MELAS (see Chapter 87) |
| Arrhythmogenic cardiomyopathy (ACM) | Scattered fibrofatty infiltration, classically of the right ventricle but also commonly involving the left ventricle; RV dilation, LV dilation, or both are common although not universal | Ventricular arrhythmias (VT, VF) early or late, reduced contractility with progressive disease; can mimic DCM | Islands of fatty replacement; fibrosis | Palmoplantar keratoderma, wooly hair in Naxos syndrome | Mutations of genes encoding proteins of the desmosome (see Table e65-1 and Fig. 65-6) | Naxos syndrome |
| Left ventricular noncompaction (LVNC) | Ratio of noncompacted to compacted myocardium increased; normal chamber dimensions varying to a DCM phenotype | Normal to reduced systolic function | Myocardium normal and ranging to findings consistent with other coexisting cardiomyopathy | Phenotype has been observed in the setting of other types of cardiomyopathy | Various cardiomyopathy genes associated but uncertain whether genetic cause or developmental defect during organogenesis; see text | |
| Infiltrative | Usually thickened walls; occasional dilation | Restrictive physiology; systolic function usually mildly reduced | Specific to type, diagnosis: amyloid, iron, glycogen storage disease, others | See RCM above | See RCM above | |
| Inflammatory | Normal or dilated without hypertrophy | Reduced systolic function | Inflammatory infiltrates | Hypereosinophilic syndrome (see text), acute myocarditis (see Chapter 67) | ||
| Ischemic | Normal or dilated without hypertrophy | Reduced systolic function | Areas of infarcted myocardium | Hypercholesterolemia, hypertension, diabetes, cigarette smoking, family history | Familial hypercholesterolemia; other heritable lipid disorders (see Chapter 45) | Familial hypercholesterolemia |
| Infectious | Normal or dilated without hypertrophy | Reduced systolic function | Specific to infection | Viral (especially acute myocarditis); protozoal (e.g., Chagas); bacterial, direct infection (e.g., Lyme disease) or from acute cellular toxicity as a result of systemic toxins (Streptococcus, gram-negatives, etc.) (see Chapter 67) | Genetic predisposition to infection and/or variable response to infective agent | |
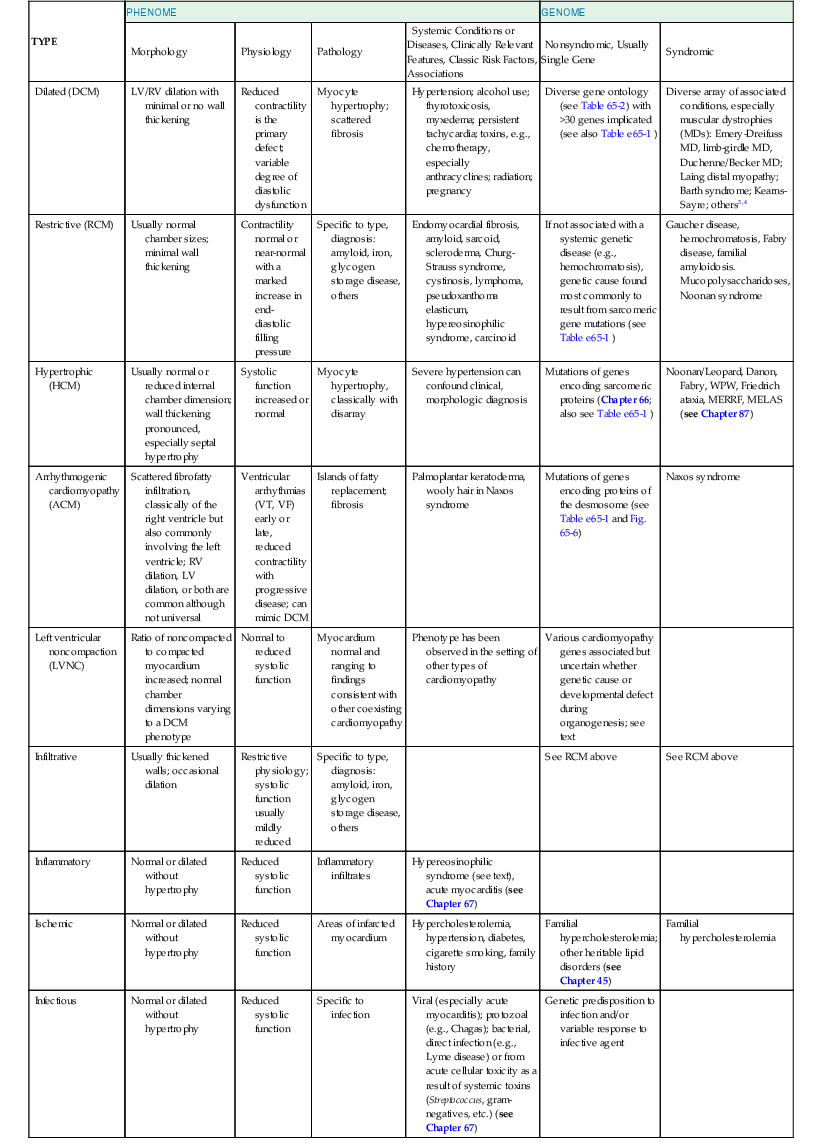
MELAS = mitochondrial encephalopathy, lactic acidosis, and strokelike symptoms; MERRF = myoclonic epilepsy associated with ragged-red fibers; RV = right ventricle; VF = ventricular fibrillation; VT = ventricular tachycardia; WPW = Wolff-Parkinson-White.
The Dilated Cardiomyopathies
DCM is characterized by a dilated left ventricle with systolic dysfunction that is not caused by ischemic or valvular heart disease. A large number of genetic causes of DCM should be considered (Table 65-2) before labeling the cardiomyopathy as “idiopathic,” which is a term that reflects our inability to make a specific diagnosis. A latent period of asymptomatic LV systolic dysfunction often occurs before the development of clinical symptoms in patients with DCM (Fig. 65-2). Patients with DCM are also at risk for ventricular arrhythmias and may occasionally initially be seen because of aborted sudden cardiac death (see also Chapter 39).
TABLE 65-2
Gene Ontology for Nonsyndromic Dilated Cardiomyopathy
| Sarcomere |
| Ion Channel |
| Cytoskeleton |
| Mitochondrial |
| Z-Disc |
| Nuclear Envelope |
| Gamma-Secretase Activity |
| Sarcoplasmic Reticulum |
| Transcription Factor |
| RNA Binding |
| Cochaperone, Heat Shock Protein |

Frequencies of mutations for each gene contributing to DCM, HCM, RCM, and ACM are provided in Table e65-1. In contrast to the many genes of diverse ontology for DCM shown here, only four sarcomere genes account for most HCM detected by clinical genetic sequencing (MYH7 and MYBPC3 account for 80%, and TNNT2 and TNNI3 account for an additional 10%).
When investigating a patient with DCM, a full history, including risk factors for coronary artery disease, should be acquired. Unless the patient is questioned in detail, the duration of symptoms may be significantly underestimated. Angina may occur, even in the absence of epicardial coronary disease, but symptoms suggestive of angina should raise the possibility of coronary artery disease, either coexistent or as a major causative factor. Patients should be questioned carefully about alcohol consumption (see Chapter 68), both present and past. If a spouse is available, that person’s input may be of great value because underinterpretation of heavy alcohol intake is common. A family history is essential, not only of symptoms suggestive of heart failure but also of sudden cardiac death, which may be referred to by the patient as “death from a massive heart attack.” Occasionally, the constellation of symptoms may allow an astute clinician to detect an uncommon cause; for example, the combination of deafness, maternally inherited diabetes, and heart failure in a relatively young patient suggests a mitochondrial cardiomyopathy.
Findings on clinical examination reflect the biventricular dysfunction present in DCM (see Chapter 23). Electrocardiography frequently reveals LV hypertrophy (LVH), nonspecific ST-T wave changes, or bundle branch block (see Chapter 23). Pathologic Q waves may be present, although their presence should raise the possibility of advanced atherosclerotic heart disease rather than primary cardiomyopathy. In advanced cases with extensive fibrosis, low-voltage limb leads may be seen.
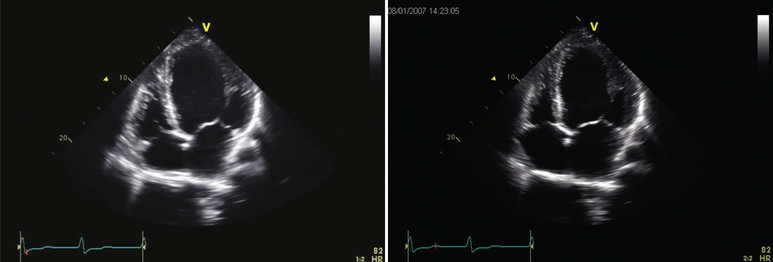
Echocardiography (see also Chapter 14) reveals biventricular dilation, which can range from mild to severe, as can LV systolic dysfunction (Fig. 65-3). LV wall thickness is usually within the normal range, but LV mass is almost invariably increased. Most commonly, global LV hypokinesis is present, but regional wall motion abnormalities may also be seen, particularly septal dyskinesis in those with left bundle branch block. Disproportionate thinning of a dyskinetic wall should raise the possibility of coronary artery disease rather than primary cardiomyopathy. Mitral and tricuspid regurgitation is frequently present and may be severe, even when the clinical examination does not reveal a loud murmur. Other than impaired leaflet coaptation, the mitral and tricuspid valves appear to be structurally normal, and structural abnormalities suggest primary valvular disease rather than cardiomyopathy. Diastolic function in DCM ranges from normal to restrictive (see also Chapter 27). A restrictive pattern is most commonly seen in patients with volume overload in “decompensated” heart failure and often improves with initiation of diuretic or vasodilator therapy.
Coronary angiography (see Chapter 20) should be considered in all patients who have risk factors for coronary artery disease or who are of an age at which this may be a causative factor. Alternatively, computed tomography (CT) coronary angiography (see Chapter 18) may be used, although it does not allow hemodynamic study, which may be useful in some patients. Because coronary artery disease is common, the functional significance of any obstructive coronary lesions found should be carefully evaluated insofar as their presence may be coincidental to DCM.
Cardiac magnetic resonance imaging (MRI) (see also Chapter 17) can be helpful in evaluating cardiomyopathies. A pattern of nontransmural delayed gadolinium enhancement in a noncoronary distribution in a dilated left ventricle suggests a nonischemic cause. Certain conditions, such as sarcoidosis, may have a rather typical appearance.6 MRI is able to evaluate the extent of myocardial fibrosis in DCM and may provide information complementary to that obtained with cardiac biopsy. Unless a specific condition is suspected, cardiac biopsy is often unrewarding in the evaluation of DCM, but it may occasionally provide an unexpected diagnosis.7 The risk for perforation during heart biopsy should be weighed against the small likelihood of finding a treatable cause with it.
Genetics of Dilated Cardiomyopathy
Despite a comprehensive evaluation, a significant proportion of patients with DCM have no obvious cause of the cardiomyopathy and are assigned a diagnosis of idiopathic DCM. Extensive family-based studies have shown that if clinical screening with an electrocardiogram (ECG) and/or echocardiogram is conducted in the first-degree family members of patients with DCM, evidence of DCM will be found in at least 20% to 35% of them, thereby establishing a diagnosis of familial DCM.8 Familial DCM is now thought to have a genetic basis of diverse ontology (see Table 65-2).9 Recent studies in families with familial DCM suggest that a genetic cause can be identified in at least 30% of cases and perhaps in as high as 40% to 50% as extrapolated from studies of individual genes or small numbers of genes in gene discovery publications (see Table e65-1 ). Even though familial DCM is now considered a genetic disease, the issue of whether idiopathic DCM has a genetic basis in cases in which there is no evidence of familial DCM has not been resolved completely.10 Patients with DCM typically have an asymptomatic phase for many years before symptomatic heart failure, an arrhythmia, or an embolic event develops later in the course of the disease (see Fig. 65-2).5 Occasionally, asymptomatic but clinically detectable DCM is discovered serendipitously during routine or preprocedure medical screening, usually prompted by subtle abnormalities on the ECG that lead to an echocardiogram. The time span needed for clinical disease to develop illustrates the remarkable ability of the myocardium to maintain normal—or close to normal—cardiac output and filling pressure for years despite easily detectable asymptomatic DCM. This principle underlies the observation that the family history is much less sensitive than clinical screening via echocardiography in detecting DCM among family members of an individual with a new diagnosis of idiopathic DCM and emphasizes the necessity of clinical screening of all first-degree family members when a new diagnosis of any cardiomyopathy has been made (Table 65-3).
). Even though familial DCM is now considered a genetic disease, the issue of whether idiopathic DCM has a genetic basis in cases in which there is no evidence of familial DCM has not been resolved completely.10 Patients with DCM typically have an asymptomatic phase for many years before symptomatic heart failure, an arrhythmia, or an embolic event develops later in the course of the disease (see Fig. 65-2).5 Occasionally, asymptomatic but clinically detectable DCM is discovered serendipitously during routine or preprocedure medical screening, usually prompted by subtle abnormalities on the ECG that lead to an echocardiogram. The time span needed for clinical disease to develop illustrates the remarkable ability of the myocardium to maintain normal—or close to normal—cardiac output and filling pressure for years despite easily detectable asymptomatic DCM. This principle underlies the observation that the family history is much less sensitive than clinical screening via echocardiography in detecting DCM among family members of an individual with a new diagnosis of idiopathic DCM and emphasizes the necessity of clinical screening of all first-degree family members when a new diagnosis of any cardiomyopathy has been made (Table 65-3).
TABLE 65-3
Summary of Guidelines for the Evaluation of Genetic Cardiomyopathies from the Heart Failure Society of America
| ABBREVIATED HFSA GUIDELINES | COMMENTS | |
| 1 | A comprehensive family history for 3 generations or more is needed with any diagnosis of cardiomyopathy. | An expert, carefully obtained family history by a skilled professional is essential and is the key first step in any genetic evaluation. |
| 2 | Clinical screening for all first-degree relatives is indicated with a new diagnosis of any cardiomyopathy that may have a genetic cause. | Because of the age-dependent penetrance, disease easily detectable by electrocardiography and echocardiography is commonly found in asymptomatic individuals. This drives the need for clinical screening of at-risk relatives. |
| 3 | Consider referral for genetic evaluation if on-site expertise is not available. | Genetic counseling, testing, and family evaluation are complex processes, so referral to centers with expertise, especially in complex or syndromic disease, should be considered. |
| 4 | In most cases genetic testing should be undertaken for the one clearly affected person in a family to facilitate family screening and management. | Selecting the optimal individual in the family for genetic testing is key. Usually this is the family member with the most definitive phenotype. Genetic testing to help establish a diagnosis in the proband may be appropriate in some circumstances. |
| 5 | Genetic and family counseling is recommended for all patients and families with cardiomyopathy. | Counseling provides updated genetic knowledge of the condition being evaluated and the probabilistic and not determinative nature of molecular genetics, including the risks and benefits of molecular genetic testing and the uncertainty and possible outcomes of results. |
| 6, 7 | Therapies based on phenotype are recommended, consistent with more general guidelines for the condition. | Drug and device guidelines for DCM, HCM, and ACM are available from the major U.S. and European organizations. |
| 8 | Implantable cardiac defibrillators should be considered for primary prevention, depending on the genotype and phenomic data, including the family history, before the usual criteria for ICD implantation may be met. | Mutations in some genes that cause DCM (e.g., LMNA) may be associated with considerable risk for sudden cardiac death before the ejection fraction falls below 35%. See discussions in the guideline documents. |
Data from Hershberger RE, Lindenfeld J, Mestroni L, et al: Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail 15:83, 2009.
TABLE e65-1
Genes Reported to Harbor Rare Variants in Association with Nonsyndromic Cardiomyopathy
| GENE* | PROTEIN | FUNCTION | OMIM | DCM† | RCM† | ACM† | LVNC† | HCM† |
| TTN | Titin | Sarcomere structure/extensible scaffold for other proteins | 188840 | 18-25% | CR | |||
| LMNA | Lamin A/C | Structure/stability of inner nuclear membrane; gene expression | 150330 | 6% | CR | |||
| MYH7 | Beta-myosin heavy chain | Sarcomeric protein; muscle contraction | 160760 | 5% | CR | CR | 40% | |
| TNNT2 | Cardiac troponin T | Sarcomeric protein; muscle contraction | 191045 | 4% | CR | CR | 5% | |
| SCN5A | Sodium channel | Controls sodium ion flux | 600163 | 3% | ||||
| MYH6 | Alpha-myosin heavy chain | Sarcomeric protein; muscle contraction | 160710 | 3% | CR | |||
| MYPN | Myopalladin | Sarcomeric protein, Z-disc | 608517 | 2% | ||||
| MYBPC3 | Myosin-binding protein C | Sarcomeric protein; muscle contraction | 600958 | 2% | CR | 40% | ||
| RBM20 | RNA binding protein 20 | RNA binding protein of the spliceosome | 2% | |||||
| ANKRD1 | Ankyrin repeat domain–containing protein 1 | Cardiac ankyrin repeat protein (CARP); localized to the myopalladin/titin complex | 609599 | 2% | ||||
| TMPO | Thymopoietin | Also LAP2, a lamin-associated nuclear protein | 188380 | 1% | ||||
| LAMA4 | Laminin a-4 | Extracellular matrix protein | 600133 | 1% | ||||
| VCL | Metavinculin | Sarcomere structure; intercalated discs | 193065 | 1% | ||||
| LDB3 | Cypher | Cytoskeletal assembly; targeting/clustering of membrane proteins | 605906 | 1% | CR | |||
| TCAP | Titin-cap or telethonin | Z-disc protein that associates with titin; aids sarcomere assembly | 604488 | 1% | ||||
| PSEN1/2 | Presenilin 1/2 | Transmembrance proteins, gamma-secretase activity | 104311/ 600759 | 1% | ||||
| MURC | Muscle-restricted coil | Z-disc protein; aids sarcomere assembly | NA | 1% | ||||
| ACTN2 | Alpha-actinin-2 | Sarcomere structure; anchor for myofibrillar actin | 102573 | <1% | CR | |||
| CRYAB | Alpha B crystalin | Cytoskeletal protein | 123590 | <1% | ||||
| TPM1 | Alpha-tropomyosin | Sarcomeric protein; muscle contraction | 191010 | <1% | CR | CR | 2% | |
| ABCC9 | SUR2A | Kir6.2 regulatory subunit, inwardly rectifying cardiac KATP channel | 601439 | <1% | ||||
| ACTC1 | Cardiac actin | Sarcomeric protein; muscle contraction | 102540 | <1% | CR | CR | <1% | |
| PDLIM3 | LIM domain protein 3 | Cytoskeletal protein | 605889 | <1% | ||||
| ILK | Integrin-linked kinase | Intracellular serine-threonine kinase; interacts with integrins | 602366 | <1% | ||||
| TNNC1 | Cardiac troponin C | Sarcomeric protein; muscle contraction | 191040 | <1% | CR | CR | ||
| TNNI3 | Cardiac troponin I | Sarcomeric protein, muscle contraction; also seen as recessive | 191044 | <1% | CR | 5% | ||
| PLN | Phospholamban | Sarcoplasmic reticulum Ca2+ regulator; inhibits SERCA2 pump | 172405 | <1% | CR | |||
| DES | Desmin | DAGC; transduces contractile forces | 125660 | <1% | ||||
| SGCD | Delta-sarcoglycan | DAGC; transduces contractile forces | 601411 | <1% | ||||
| NEBL | Nebulette | Binds actin; Z-disc assembly | 605491 | <1% | ||||
| NEXN | Nexilin | Cardiac Z-disc | 613122 | <1% | ||||
| CSRP3 | Muscle LIM protein | Sarcomere stretch sensor/Z-discs | 600824 | <1% | CR | |||
| EYA4 | Eyes-absent 4 | Transcriptional coactivators (Six and Dach) | 603550 | CR | ||||
| DMD‡ | Dystrophin | Dystrophin-associated glycoprotein complex, transduces contractile force | 300377 | ? | ||||
| TAZ‡ | Tafazzin | Unknown | 300394 | CR | CR | |||
| MYL2 | Regulatory myosin light chain | Sarcomeric protein; stabilizes long helical neck of myosin head | 160781 | CR | <1% | |||
| MYL3 | Eseential myosin light chain | Sarcomeric protein; stabilizes long helical neck of myosin head | 160790 | CR | 1% | |||
| PKP2 | Plakophilin 2 | Desmosomal protein | 602861 | CR | 10%-40% | |||
| DSG2 | Desmoglein | Desmosomal protein | 125671 | CR | 10%-40% | |||
| DSP | Desmoplakin | Desmosomal protein | 125647 | CR | 5%-15% | |||
| DSC2 | Desmocollin | Desmosomal protein | 125645 | CR | 2% | |||
| JUP | Junction plakoglobin | Desmosomal protein | 173325 | CR | CR | |||
| TMEM43 | Transmembrane protein 43 | 612048 | CR | CR | ||||
| TGFB3 | Transforming growth factor-beta-3 | 190230 | CR | CR | ||||
| RYR2 | Ryanodine receptor 2 | Calcium handling for myocyte contraction | 180902 | CR | CR |
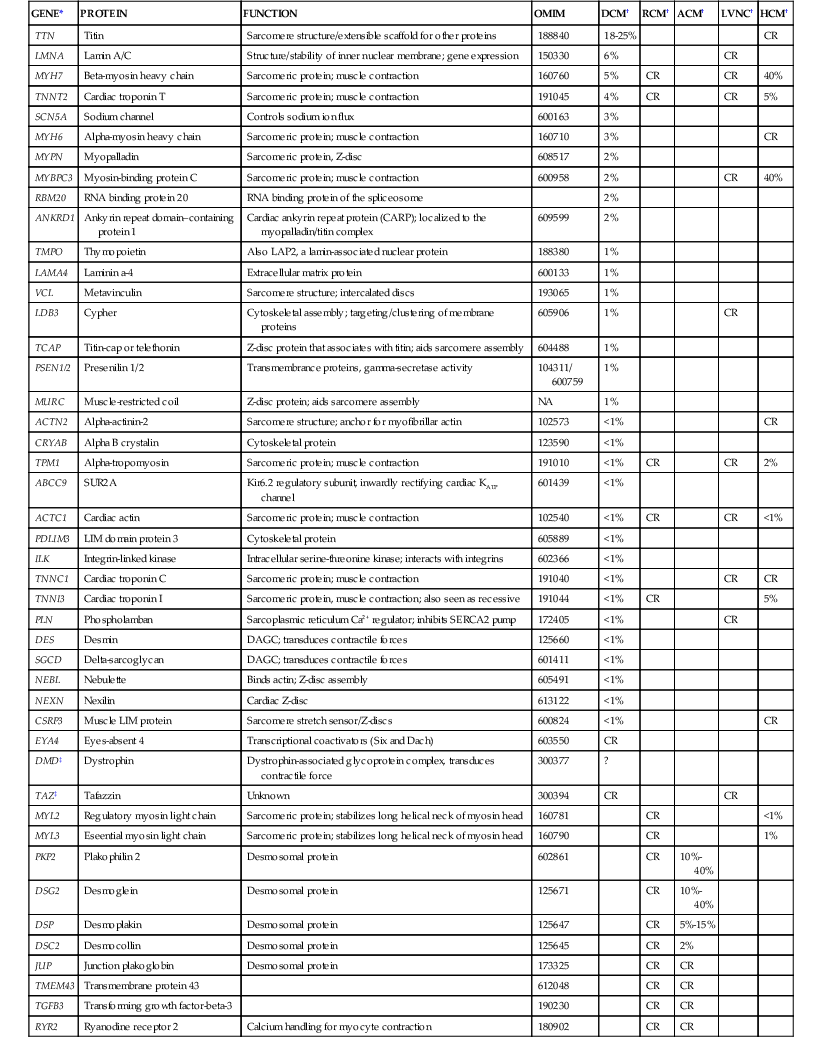
* Gene symbol.
† Percentages; the fraction of probands carrying mutations of that gene for the phenotypes shown based on primary and secondary reports.
‡ X-linked.
ATP = adenosine triphosphate; CR = case reports with insufficient data to estimate the frequency within the phenotype; NA = not available.
Molecular Genetics of Familial Dilated Cardiomyopathy
The genes shown to cause familial DCM are classified by subcellular location (gene ontology). As shown in Table 65-2, most of the implicated genes encode sarcomere, Z-disk, or cytoskeleton proteins. The broad representation of other genes encoding a wide variety of proteins demonstrates the diverse pathways that can lead to the “final phenotype” of DCM.10 Presumably other yet unknown pathways may also be relevant in the pathogenesis of DCM. More than 30 genes have been identified to cause DCM (referred to as locus heterogeneity). The diverse subcellular locations of genes implicated in DCM differentiates this form of cardiomyopathy from HCM (see also Chapter 66) and arrhythmogenic cardiomyopathy (ACM), which are caused by variants in genes encoding sarcomeric or desmosomal proteins, respectively (see Tables 65-1, 65-2, and e65-1). Recently, truncating variants in the giant scaffolding protein titin (TTN) have been suggested to cause 25% of cases of DCM.11 However, up to 3% of controls also carried TTN truncating variants, and a genome-wide strategy that demonstrated the impact and relevance of TTN truncating variants also identified some truncating variants that were not associated with DCM,12 which makes assessment of pathogenicity problematic from the standpoint of clinical genetic testing. In addition to locus heterogeneity, the molecular genetics of DCM is also characterized by allelic heterogeneity; that is, mutations commonly occur at many locations in a DCM gene, and many mutation sites in genes shown to cause both DCM and HCM are specific to that cardiomyopathy type (see Fig. e65-1 ). So-called overlap phenotypes are not uncommon, particularly for sarcomeric genes, wherein mutations that have been shown to cause DCM, HCM, and RCM may be seen in an extended pedigree. Indeed, all three phenotypes (HCM, RCM, DCM) have been reported with the same mutation in an extended family.13
). So-called overlap phenotypes are not uncommon, particularly for sarcomeric genes, wherein mutations that have been shown to cause DCM, HCM, and RCM may be seen in an extended pedigree. Indeed, all three phenotypes (HCM, RCM, DCM) have been reported with the same mutation in an extended family.13
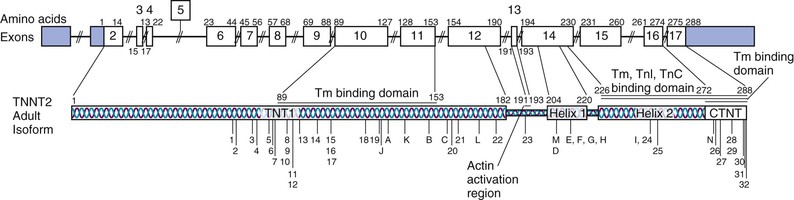
Clinical Genetics of Familial Dilated Cardiomyopathy
Familial DCM is characterized by a relative unitary final phenotype9 of “generic” DCM. That is, for almost all genes implicated in DCM, there is no unique or distinguishing genotypic or phenotypic features that have been associated with specific gene mutations.8 The only general phenotypic variation that has been noted8,10 is “DCM with prominent conduction system disease,” a phenotype that is observed in all cases of lamin A/C (LMNA) DCM and in some cases of sodium channel (SCN5A) and desmin (DES) DCM (see Table e65-1). Occasionally, a clinically mild muscular dystrophy phenotype can be identified in patients with LMNA cardiomyopathy and a new diagnosis of DCM, but in most cases the muscular dystrophy has been identified in a neuromuscular clinic, with DCM being an incidental finding at the time of evaluation. Regardless of the setting, when a new diagnosis of idiopathic DCM is made, vigilance in detecting syndromic disease is essential, with particular attention being directed to neuromuscular phenotypes.
Most familial DCM is transmitted via autosomal dominant inheritance, with the offspring of a mutation carrier having a 50% chance of inheriting the mutation. Autosomal recessive disease has been reported, particularly in consanguineous families. X-linked DCM resulting from mutations in the gene for Duchenne muscular dystrophy (DMD) in patients without any findings of muscular dystrophy has been reported both in males and in carrier females, although the prevalence of DMD-DCM in cohorts of patients with idiopathic DCM has not been studied systematically. Mitochondrial DCM has also been reported, particularly in the setting of syndromic disease.3
Familial DCM is characterized by age-dependent penetrance, which means that an individual harboring a DCM-causing allele will manifest evidence of the DCM phenotype with increasing age.8,10 Most genetic DCM becomes evident in the fourth to seventh decades, although DCM occurring in adolescence, childhood, or infancy is not uncommon. Variations in the age at onset of DCM are common across families with mutations in the same DCM gene, at times marked, and even in family members of an extended pedigree with the same mutation. Penetrance in familial DCM is commonly incomplete; that is, an individual with a disease-causing allele may not manifest any aspect of the disease phenotype. Also, expression is variable in that the clinical features, the phenotype, can vary significantly between individuals in the same family or between families with the same mutation. Both incomplete penetrance and variable expressivity confound the assessment of familial DCM in family pedigrees. This is particularly relevant for a newly discovered or novel candidate mutation in a family because full segregation of the candidate mutation with the disease phenotype in one or more extended families is one of the most powerful means of determining the pathogenicity of such variants.14
Incomplete penetrance and variable expressivity at times result in marked phenotypic variability within and between families with DCM, even with the same mutation. The explanation for this phenomenon is not clear. Both environmental and genetic factors have been postulated and range from intrinsic (e.g., hypertension) and extrinsic phenomic components (e.g., toxins, viruses, adverse or favorable drug exposure) to a combination of various genomic variants resulting in a different genetic milieu (e.g., a “second hit” from a second mutation in a different disease gene, risk alleles in the same or other relevant DCM pathways, variability in epigenetics or gene expression, and others).9,15
Allelic heterogeneity, in which mutations in one gene can give rise to different and distinct phenotypes seemingly unrelated to one another (see Fig. e65-1), is also observed with some DCM genes, and knowledge of these allelic variants can be critical when considering a genetic diagnosis of DCM.3 One of the most remarkable examples is LMNA, which encodes the proteins lamin A and lamin C, key components of the inner nuclear membrane. For example, mutations in LMNA cause a distinctive DCM phenotype in which conduction system disease and arrhythmia occur before the onset of DCM. Mutant lamin proteins also cause a variety of syndromic diseases spanning striated muscle, adipose, nerve, and vascular tissues. These phenotypes, collectively termed the laminopathies, include skeletal myopathies (autosomal dominant Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy type 1B, and others [see Chapter 87]), lipodystrophy syndromes, peripheral neuropathy, and accelerated aging syndromes, most notably Hutchinson-Gilford progeria.16
Approach to Clinical Genetic Evaluation, Including Genetic Testing
Guidelines for evaluation and clinical genetic testing for DCM, applicable to all cardiomyopathies with a possible genetic cause (see Table 65-3), include a comprehensive three- to four-generation search of the family history for any evidence of any type of cardiomyopathy, muscular dystrophy, or other evidence of syndromic disease that may have a cardiomyopathy component.4,17 However, as noted earlier, even if obtained by a skilled professional, the family history might be negative because DCM may be asymptomatic in family members. Accordingly, clinical screening of all first-degree relatives is essential, including a history, physical examination, ECG, and echocardiography at a minimum. If evidence of DCM is identified in a relative, screening of that relative’s first-degree relatives is indicated (stepwise or cascade clinical screening). Genetic testing, within the context of genetic counseling, is indicated with any evidence of familial disease because identification of a disease-associated mutation (in one or more clearly affected family members) can permit molecular genetic testing of other preclinical, but at-risk family members and thereby aid in their risk stratification. Those who test negative for the family mutation should have a significantly reduced risk for the development of DCM, whereas those with a family DCM mutation should undergo enhanced clinical screening to detect early disease, with the rationale that early intervention, usually with angiotensin-converting enzyme (ACE) inhibitors or beta blockers, may delay or prevent progression of the disease.
Genetic testing is now conducted by next-generation sequencing in panels of DCM genes ranging from 20 to 30 or more. Pan-cardiomyopathy panels now also contain more than 50 genes, and their competitive cost structure suggests that large test panels will quickly become the norm.18 Genetic testing should always be conducted within the context of genetic counseling, the goals of which are to review the genetic inheritance patterns and clinically relevant facts regarding idiopathic and familial DCM and ensure that a comprehensive family history has been completed and properly interpreted, including identification of at-risk relatives. Counseling is also essential to provide information regarding the risks, benefits, and limitations of clinical genetic testing, including the possible consequences of uncertain or inclusive results or the discovery of heritable disease and its potential psychological implications.8,10 These processes are time-consuming and require specialized knowledge, and guidelines suggest that referral of patients to individuals or centers with experience should be considered if local resources for their completion are not available.4
The recommendation for genetic testing recognizes that with the greater number of genes being tested in pan-cardiomyopathy panels, a greater number of variants of unknown or uncertain significance may be encountered.18 Clinicians ordering clinical genetic testing must understand this concept and be prepared to deal with this reality as the results become available. The emergence of next-generation sequencing of panels of genes has fueled an extremely active period for reevaluation of testing strategies, including approaches to interpreting large numbers of variants.14,19 All of this will require careful, comprehensive translational research to understand the optimal testing strategies, including large databases of disease-associated variants.
Therapy for Dilated Cardiomyopathy
Therapy for DCM is similar to that for all types of systolic dysfunction (see Chapter 25). As with all heart failure patients, avoidance of excessive dietary sodium is crucial. Beta-blocking drugs and ACE inhibitors/angiotensin receptor blockers (ARBs) are the mainstay of therapy to prevent progressive disease, even in the absence of symptoms, and diuretics are the cornerstone of therapy to reduce peripheral edema and pulmonary congestion in those with symptomatic disease, with the addition of aldosterone antagonists (spironolactone and eplerenone) in more advanced cases. Ivaradibine, a selective heart rate–lowering drug approved in Europe, may be added for patients who have a suboptimal heart rate–lowering effect with beta blockade or who cannot tolerate beta-blocking agents. Attention should be paid to treatment of atrial arrhythmias (see Tachycardia-Induced Cardiomyopathy, later). In selected patients, cardiac resynchronization therapy should be considered, sometimes even early in the course of the disease, and in those with advanced disease, referral for a ventricular assist device or cardiac transplantation may be needed (see also Chapters 28 and 29).
Alcoholic and Diabetic Cardiomyopathies
Excessive alcohol intake is cardiotoxic and may be manifested as DCM (considered in detail in Chapter 68). Unique features worthy of stressing include the fact that it rarely occurs without a history of drinking at least 90 g of alcohol daily for at least 5 years, which represents a minimum of at least eight standard drinks daily.20 The condition may be reversible with abstinence from alcohol but progresses if the patient continues to drink. The importance of obtaining as accurate an alcohol history as possible cannot be overstressed. In addition to its role in DCM, heavy alcohol use is associated with hypertension, which can aggravate DCM and may be refractory to treatment.
The existence of a specific diabetic cardiomyopathy independent of the effect of diabetes on the vasculature is debated, both in terms of its existence and, among those who believe it to exist, in the form that it takes.21 Subtle abnormalities in both systolic and diastolic function do seem to be prevalent in diabetic patients, but their clinical relevance to the development of overt disease is unclear (see Chapter 61).
Arrhythmogenic Cardiomyopathy
ACM is a genetically determined cardiomyopathy characterized by fibrofatty replacement of the myocardium. Formerly called “arrhythmogenic right ventricular dysplasia/cardiomyopathy,” it is better termed ACM because it is now recognized that biventricular involvement occurs in up to 50% of cases and that a small proportion of cases affect predominantly the left ventricle (Fig. 65-4). The disorder is conceptualized as having three stages: an early subclinical phase in which imaging studies are negative but during which sudden cardiac death can still occur; next, a phase in which (usually) right ventricular (RV) abnormalities are obvious without any clinical manifestation of RV dysfunction but with the development of symptomatic ventricular arrhythmia; and finally, progressive fibrofatty replacement and infiltration of the myocardium leading to severe RV dilation and aneurysm formation and associated right-sided heart failure (Fig. e65-2 ). LV dilation and failure may also arise at this stage or may occur later (sometimes referred to as phase 4).22
). LV dilation and failure may also arise at this stage or may occur later (sometimes referred to as phase 4).22
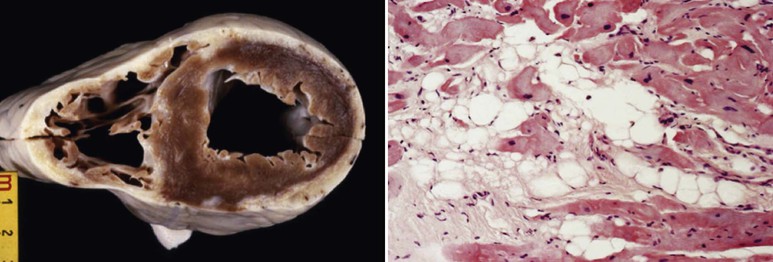
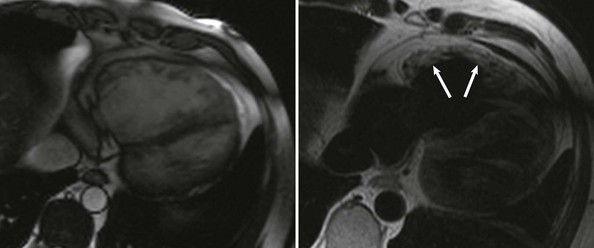
The electrical manifestations of ACM are a reflection of the pathologic disturbance. In the early stage, slow conduction and electrical uncoupling may lead to a fatal arrhythmia. As the disease progresses, fibrofatty infiltration results in inhomogeneous activation and a further delay in conduction. The predominant site of RV involvement is often the “triangle of dysplasia,” an area involving the RV outflow tract, an area below the tricuspid valve, and the RV apex; this is the most common area of RV thinning, regional dilation, and aneurysm formation. This anatomic area and its abnormalities in ACM are responsible for the typical ECG appearance during sinus rhythm and a typical monomorphic ventricular tachycardia (VT) characterized by left bundle branch block morphology with a superior axis23 (Fig. 65-5).
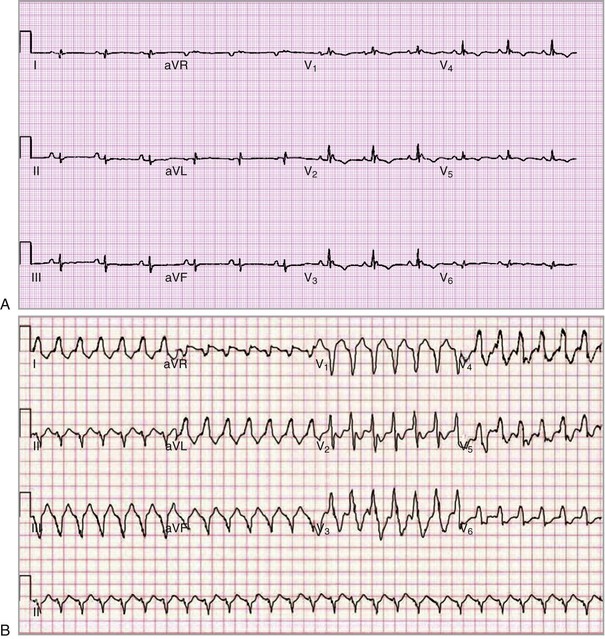
An intriguing postulate to explain the progressive nature of ACM and the predilection for the pathology to affect the right ventricle comes from the observation that the prevalence of sudden cardiac death may be higher in athletes with ACM than in sedentary patients. This has led to the hypothesis that the right ventricle, being thinner than the left ventricle, is more susceptible to mechanical stretch, particularly during exercise. Mechanotransduction, or conversion of mechanical stimuli to biochemical intracellular signals, may further increase dysfunction at the cellular level.
Genomic Cause of Arrhythmogenic Cardiomyopathy
Unlike genetic DCM, which has a final common phenotype despite its extensive locus heterogeneity, ACM is driven by molecular genetic alterations in genes encoding proteins that are key for cell-to-cell adhesion (Fig. 65-6).24 Extensive work over the past decade has implicated genes encoding the desmosome, one of three key components of the intercalated disc, the end-to-end connection between ventricular myocytes24 in the pathogenesis of ACM. In addition to desmosomes, the intercalated disc includes gap junctions mediating small-molecule communication. Mechanical coupling is mediated through the desmosome and adherens junctions (see Chapter 21), and disruptions of desmosomal proteins have been associated with ACM. The classic hallmark of ACM, fibrofatty replacement, is now understood to be related to aberrant Wnt signaling of desmosomal proteins, as well as direct plakoglobin signaling, which transforms myocytes into adipocytes with disease progression.24
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


