The Biology of Restenosis
Eric Van Belle
Christophe Bauters
Michel E. Bertrand
Overview
Neointimal thickening, also referred to as neointimal hyperplasia, occurs in response to experimental arterial injury with a balloon catheter. This process, which is mainly the consequence of a growth response of the smooth muscle cells (SMCs), is maximal 1 to 4 weeks after the initial injury. It occurs in different vascular beds (coronary arteries, peripheral arteries) and in various animal species, including rats, rabbits, and pigs. A marked hyperplastic response can be observed after a single injury in normal vessels but also in animal models in which a first injury combined with a hypercholesterolemic diet has induced a stenosis that can then be dilated with an angioplasty balloon catheter. Neointimal formation involves the activation, proliferation, and migration of SMCs and the production of extracellular matrix (ECM).
The factors that control neointimal thickening include growth factors, hormonal factors, and mechanical factors. Growth factors released at the site of injury play a major role in the response of SMCs to balloon injury and include platelet-derived growth factor (PDGF), basic fibroblastic growth factor (bFGF), transforming growth factor-β (TGF-β), and insulin-like growth factor-1 (IGF-1). Hormonal factors include angiotensin II, serotonin, and endothelin. Mechanical factors include trauma related to interventional devices.
Delinquent reendothelialization has been shown to have a permissive, if not facilitatory, effect on SMC proliferation. This inverse relationship has been attributed to certain functions of the endothelium, including barrier regulation of permeability, thrombogenicity, and leukocyte adherence, as well as to growth-inhibitory molecules. More-recent studies have confirmed this notion by demonstrating that administration of endothelial cell mitogens may facilitate endothelial cell regeneration, reduce neointimal thickening, and promote recovery of endothelial dysfunction after balloon injury.
Neointimal hyperplasia is not the sole mechanism leading to lumen renarrowing after balloon angioplasty. A change in external vessel size, namely “arterial remodeling,” also plays a role in this process. Studies performed in animals and humans have established the potential for “constrictive remodeling” to reduce vessel wall area after angioplasty, which indirectly narrows the vessel lumen and thus contributes to restenosis after balloon angioplasty.
Histologic findings after stent deployment in animal models and analyses of specimens retrieved from patients have established that vessel remodeling is virtually abolished by stent implantation and that in-stent restenosis is purely due to intimal hyperplasia.
Recently studies have shown that strategies interfering with proliferation and migration of vascular cells (endovascular brachytherapy, stents eluting antiproliferative drugs) might interfere with intimal hyperplasia in animal models and are useful for treating or preventing in-stent restenosis in humans. Histologic findings have suggested, however, that this benefit is achieved at the price of a delayed vascular healing. In that regard, it is interesting that strategies designed to promote endothelialization of the stent struts reduce the thickness of the neointima and are associated with an accelerated vascular healing in animal models. Still, the potential benefit of this attractive approach is unknown in humans.
Introduction
The treatment of coronary artery disease is one of the paramount problems in cardiology. Percutaneous transluminal coronary angioplasty (PTCA) has become a well-established technique for myocardial revascularization of patients with coronary artery disease (1,2,3). An estimated 657,000 PTCA procedures were performed on 640,000 patients in 2002 in the United States. From 1987 to 2002 the number of procedures increased 324% (4). For many years, the use of PTCA was limited by restenosis, which occurred in 30% to 60% of patients despite performance of a successful procedure (5,6,7,8).
During the last two decades, numerous agents have been used in an attempt to prevent restenosis (9). Despite positive results in animal models, no pharmacologic therapy has been found to significantly decrease the risk of restenosis in humans. These apparent discrepancies between animal models and the clinical situation are probably related mainly to an incomplete understanding of the mechanisms of restenosis in humans as well as in animal models.
During the last few years, important experimental and clinical studies have allowed a better understanding of the various processes that occur after PTCA and that may lead to restenosis. The understanding of the role of “constrictive vessel remodeling” (10,11) led to the development of coronary stenting, which is associated with a 50% decrease in restenosis rate compared to balloon angioplasty (8,12,13). More recently it was demonstrated that local delivery of antiproliferative agents (radiation, drugs) could interfere with intimal hyperplasia (8,14,15,16,17,18) in animal models and further decrease the restenosis rate after stent implantation in humans (8,19,20,21,22,23).
The purpose of this chapter is to review the information relevant to the response of the vascular wall to injury. The focus is on four major processes: neointimal hyperplasia, vessel remodeling, thrombosis, and endothelial regeneration. The mechanisms of these processes, as well as their relative contributions to lumen renarrowing in animals and humans, are reviewed in an attempt to reconcile results for animal models of restenosis with the clinical situation.
 FIGURE 102.1. Experimental balloon denudation of rabbit aorta. A: Noninjured artery. B: Immediately after balloon denudation, complete deendothelialization is apparent. C: Immediately after balloon denudation, a break is also seen in the internal elastic membrane. D: Twenty-eight days after balloon denudation, neointimal thickening is observed. Arrow indicates the internal elastic lamina. |
Neointimal Hyperplasia
Neointimal Hyperplasia after Experimental Arterial Injury
During the last 10 to 15 years, many studies have described the process of neointimal thickening—also known as neointimal hyperplasia—that occurs in response to experimental arterial injury with a balloon catheter (24,25,26,27,28,29,30,31) (Fig. 102.1). This process is due largely to growth of the SMCs and is maximal 1 to 4 weeks after the initial injury (Fig. 102.2). Neointimal thickening occurs both in coronary arteries and in peripheral arteries and is found in various animal species, including rats, rabbits, and pigs (24,31,32,33). A single injury in a normal vessel can produce a marked hyperplastic response (24,31,34). Neointimal hyperplasia is also observed in animal models fed a hypercholesterolemic diet after a first injury to induce a stenosis that can then be dilated with an angioplasty balloon catheter (double-injury model) (27,28). Neointimal formation involves several steps: SMC activation, proliferation, and migration and the production of ECM.
Proliferation and Migration of Smooth Muscle Cells
Activation of SMCs is associated with a shift from a contractile to a synthetic phenotype (35,36) and leads to proliferation, migration, and synthesis of ECM. Proliferation of medial SMCs is evident 24 hours after experimental balloon injury and continues for at least 2 weeks (26). At least 20% to 40% of
medial SMCs are activated and enter the cell cycle between 24 hours and 3 days after balloon denudation (37). In vivo animal studies demonstrated the extent to which cell-cycle events are temporally coordinated after balloon injury (38). These cells then migrate to the intima through breaks in the internal elastic membrane. SMCs are observed on the luminal side of the internal elastic lamina 4 days after injury to rat arteries (24). Many of these neointimal cells continue to proliferate for several cycles, but nearly half of the migrating cells do not synthesize DNA (37). Proliferation and migration should thus be considered as two distinct mechanisms leading to neointimal thickening; as discussed later, some factors may affect SMC migration but have no effect on SMC proliferation, and vice versa (25).
medial SMCs are activated and enter the cell cycle between 24 hours and 3 days after balloon denudation (37). In vivo animal studies demonstrated the extent to which cell-cycle events are temporally coordinated after balloon injury (38). These cells then migrate to the intima through breaks in the internal elastic membrane. SMCs are observed on the luminal side of the internal elastic lamina 4 days after injury to rat arteries (24). Many of these neointimal cells continue to proliferate for several cycles, but nearly half of the migrating cells do not synthesize DNA (37). Proliferation and migration should thus be considered as two distinct mechanisms leading to neointimal thickening; as discussed later, some factors may affect SMC migration but have no effect on SMC proliferation, and vice versa (25).
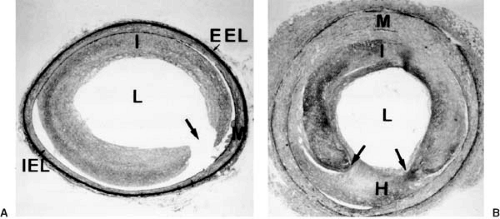 FIGURE 102.2. Experimental balloon angioplasty of an iliac artery in the hypercholesterolemic rabbit model. A: Immediately after balloon angioplasty, rupture (arrow) of the atherosclerotic lesion is seen. B: Twenty-eight days after balloon angioplasty, the site of plaque rupture is “grouted in” by neointimal hyperplasia (H), maximal at the site of plaque rupture (elastic tissue stain). EEL, external elastic lamina; I, intima; IEL, internal elastic lamina; L, lumen; M, media. |
It should also be noted that an alternative to the predominant hypothesis that all SMCs of the media can undergo phenotypic modulation (35,36) is the concept that a predisposed SMC subpopulation is responsible for the production of intimal thickening. This concept is based on the seminal observation of Benditt and Benditt (39) that human atheromatous plaques have the features of a monoclonal or an oligoclonal lesion and implies that under normal conditions, SMCs are phenotypically heterogeneous (40).
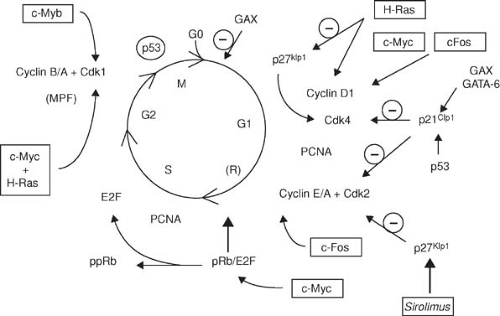 FIGURE 102.3. Simplified scheme of the cell cycle. Cell-cycle progression depends on the expression and activation of specific enzymes, termed cyclin-dependent kinases (Cdk’s), which combine with their regulatory subunits, the cyclins. Cyclin-dependent kinase inhibitors (p27Kip1, p21Cip1) bind to and inhibit the activation of Cdk/cyclin complexes. Also shown are the gene products of protooncogenes (in boxes), which act as transcription factors that influence the expression of specific cell-cycle regulatory genes and the site of action of sirolimus. E2F, transcription factor E2F; MPF, mitosis-promoting factor; PCNA, proliferating-cell nuclear antigen; ppRb hypophosphorylated retinoblastoma gene product; pRb, retinoblastoma gene product. (From Braun-Dullaeus RC, Mann MJ, Dzau VJ. Cell cycle progression—new therapeutic target for vascular proliferative disease. Circulation 1998;98:82–89 .) |
Finally, other studies suggest that some SMCs involved in intimal hyperplasia may originate from other sources, including fibroblast from the adventitia (41), endothelial cells (42), and/or circulating bone marrow–derived cells (43,44). Although studies suggest that in some circumstances these cells may represent 40% of intimal cells (45), their precise role in intimal hyperplasia is unclear.
Cell Cycle and Vascular Smooth Muscle Cell Proliferation
Growth factors and cytokines share a final proliferative signaling pathway: the cell cycle (Fig. 102.3). Quiescent (G0) cells




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
