Chapter 41 The Adrenal Glands
History
The adrenal glands were first described by the Italian anatomist Bartolomeo Eustachi in 1563. The German comparative anatomist Albert von Kölliker (1817-1905), who noted the presence of the adrenals in a number of vertebrate species, is credited with first identifying two distinct portions of the adrenal gland, the cortex and the medulla. Although Thomas Addison described the clinical features of primary adrenal failure in 1855, it was not until almost a century later that the adrenal hormones were fully isolated and characterized. Adrenaline (or epinephrine) was first isolated from adrenal extract at the turn of the century. Steroid hormones were crystallized from cortical extract (cortin) by Swiss and American investigators in the 1930s, but their highly similar chemical structures made isolation of the individual compounds challenging. Edward Kendall, Tadeus Reichstein, and Philip Hench jointly received the 1950 Nobel Prize in Physiology or Medicine for their groundbreaking work on the adrenocortical hormones. The Austrian-born endocrinologist Hans Selye first described the stress response in mammals in 1936 and made major contributions to the understanding of the hypothalamic-pituitary-adrenal (HPA) axis. Roger Guillemin, Andrew Schally, and Rosalyn Yalow were awarded the Nobel Prize in Physiology or Medicine in 1977 for characterizing the peptide hormones of the brain that underlie the HPA axis as we now understand it.1,2
Anatomy and Embryology
General and Developmental Aspects
The adrenal glands are paired, mustard-colored structures positioned superior and slightly medial to the kidneys in the retroperitoneal space (Fig. 41-1). They are flattened and roughly pyramidal (right) or crescent-shaped (left), weighing approximately 4 g each. The adrenals are among the most highly perfused organs in the body, receiving 2000 mL/kg/min of blood, after only the kidney and thyroid. In most respects, the cortex and medulla can be considered as two completely distinct organs that happen to colocalize during development. The two portions have disparate embryologic origins. The primordial cortex arises from the coelomic mesodermal tissue near the cephalic end of the mesonephros during the fourth to fifth week of gestation. Biosynthetic activity can be detected as early as the seventh week. Cortical cell mass dominates the fetal adrenal at 4 months of development and steroidogenesis reaches is maximum during the third trimester. The adrenal medulla arises from the ectodermal tissues of the embryonic neural crest. It develops in parallel with the sympathetic nervous system, beginning in the fifth to sixth week of gestation. From their original position adjacent to the neural tube, neural crest cells migrate ventrally to assume a para-aortic position near the developing adrenal cortex. There, they differentiate into chromaffin cells that make up the adrenal medulla.
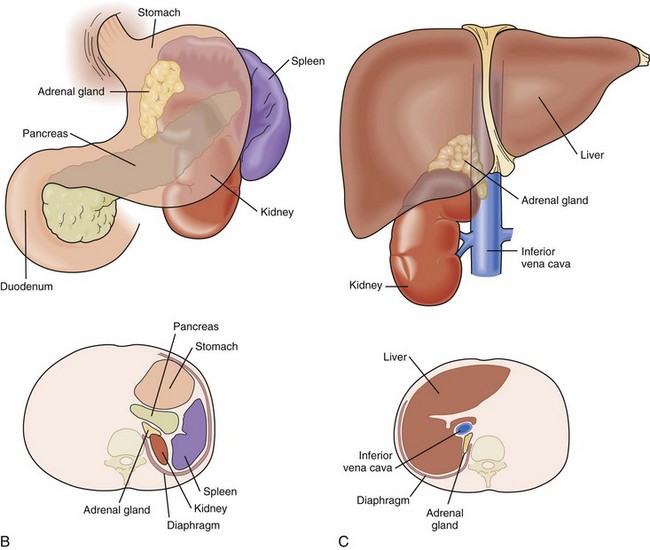
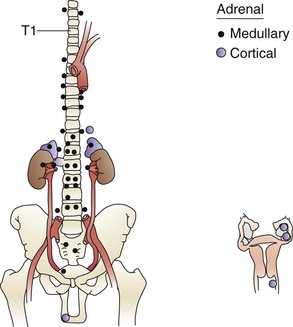
This course of embryologic development yields certain surgically relevant sequelae. Both cortical and medullary tissue can be found at extra-adrenal sites (Fig. 41-2). The range of potential sites is wider for chromaffin tissue than for cortical tissue. Pheochromocytomas may arise in extra-adrenal sites more commonly than previously believed (see later). When extra-adrenal, pheochromocytomas are also termed paragangliomas.
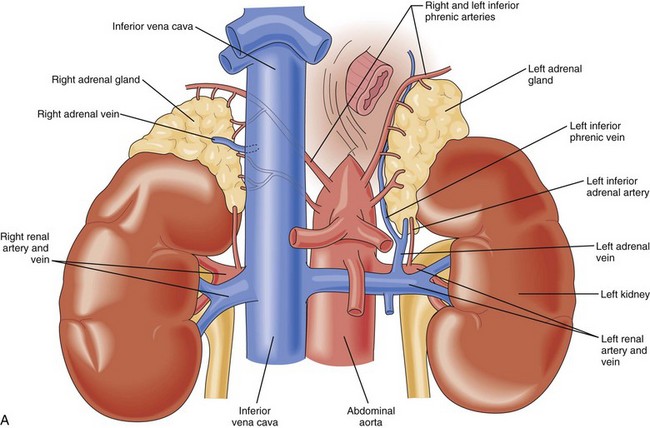
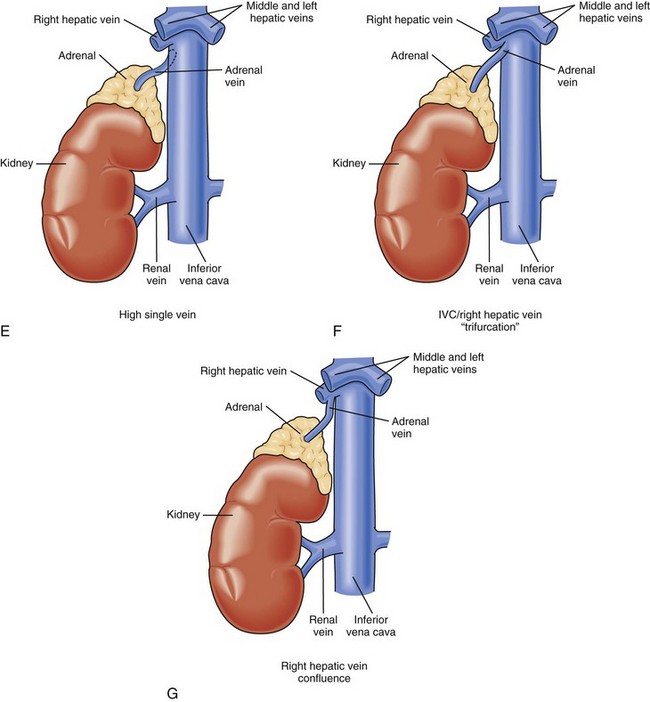
Vasculature
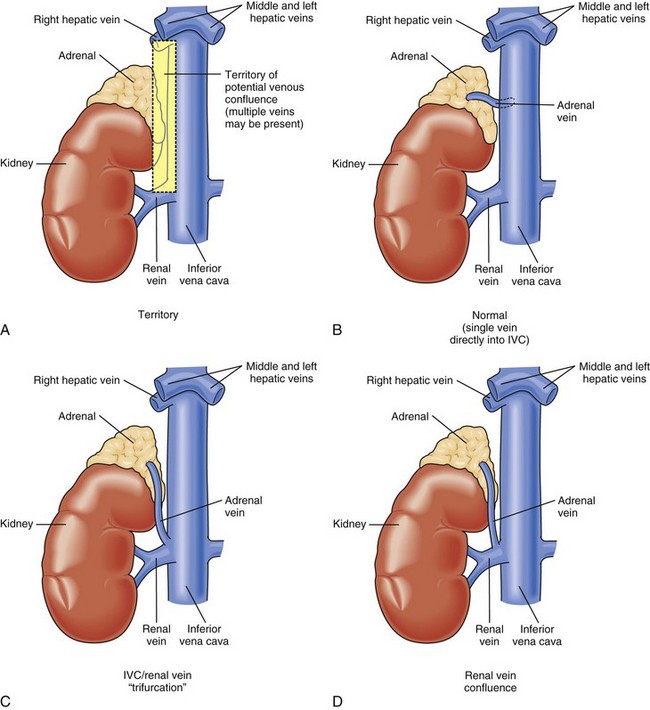
Knowledge of the macroscopic vascular anatomy of the adrenal glands is essential to proper surgical management. It is important to conceptualize that although the arterial supply is diffuse, the venous drainage of each gland is usually solitary. The arterial supply arises from three distinct vessels—the superior adrenal arteries from the inferior phrenic arteries, small middle adrenal arteries from the juxtaceliac aorta, and inferior adrenal arteries from the renal arteries. Of these, the inferior is the most prominent and is commonly a single identifiable vessel. The left adrenal vein is approximately 2 cm long and drains into the left renal vein after joining the inferior phrenic vein. The right adrenal vein is typically as short as it is wide (0.5 cm) and drains directly into the vena cava. This configuration presents a surgical challenge that will be discussed in more detail later in this chapter. In up to 20% of individuals, the right adrenal vein may drain into an accessory right hepatic vein or into the vena cava, at or near the confluence of the vein. Vigilance about this variant and others (Fig. 41-3) may reduce the likelihood of intraoperative venous hemorrhage during a right adrenalectomy.
Normal Histopathology
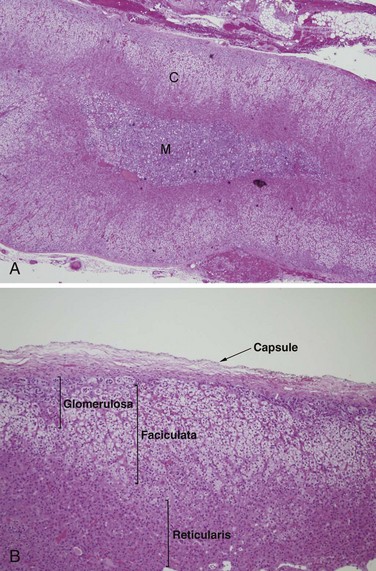
The cortex is approximately 2 mm thick and comprises more than 80% of the mass of the gland. It is made up of three layers (Fig. 41-4). The outer zona glomerulosa is a thin layer of relatively small cells with moderately eosinophilic, lipid-poor cytoplasm. It has an undulating inner border and normally does not form a complete circumferential layer. Most of the adrenal cortex is formed by the zona fasciculata, a middle layer composed of long radial columns of large, clear, lipid-laden cells. The inner zona reticularis is made up of small nests of compact eosinophilic cells. The adrenal medulla consists of clusters and short cords of chromaffin cells, which are large, polyhedral, and packed with basophilic secretory granules. Catecholamines within these granules yield a brown-colored reaction when treated with chromium salts, giving the cells their name. In contrast to the cortex, the adrenal medulla is richly endowed with autonomic nerve fibers and ganglion cells. Sympathetic fibers synapse directly with the chromaffin cells, constituting an interface between the nervous and endocrine systems.
Biochemistry and Physiology
Adrenal Steroid Biosynthesis
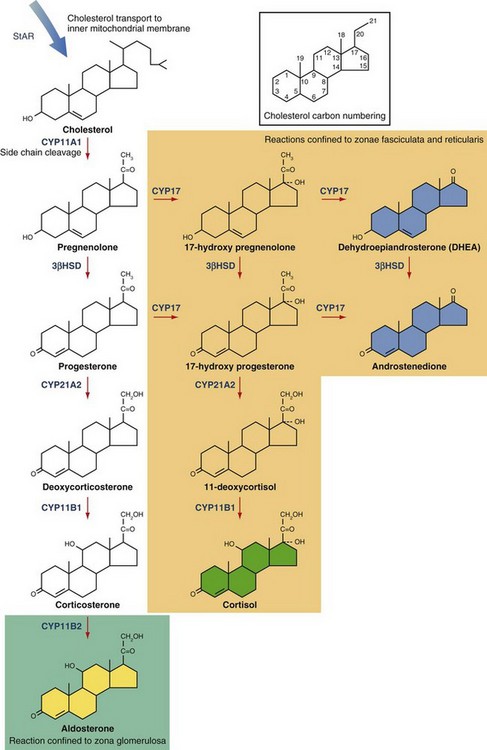
Adrenal steroid biosynthesis begins with the transport of cholesterol to the inner mitochondrial membrane by the steroidogenic acute regulatory protein (StAR; Fig. 41-5). Cholesterol then undergoes a series of oxidative reactions catalyzed predominantly by membrane-associated enzymes belonging to the cytochrome P450 (CYP) family. Cleavage of the cholesterol side chain yields the hormonally inactive compound pregnenolone, the immediate precursor to the adrenal steroid hormones. Serial oxidation by CYP17 converts pregnenolone and progesterone into the major adrenal sex steroids, dehydroepiandrosterone (DHEA) and androstenedione. Additional enzymatic steps confined to the gonads generate testosterone, estrone, and estradiol from androstenedione. Oxidation of 17-hydroxypregnenolone by 3β-hydroxysteroid dehydrogenase (3β-HSD), followed by action of CYP21A2 and CYP11B1 yields cortisol, the active glucocorticoid hormone in humans. Aldosterone is generated by the oxidation of corticosterone by CYP11B2 within the zona glomerulosa. CYP17 expression is confined to the zona fasciculata and zona reticularis, accounting for the synthesis of glucocorticoids and adrenal sex steroids in these regions.
Catecholamine Biosynthesis and Physiology
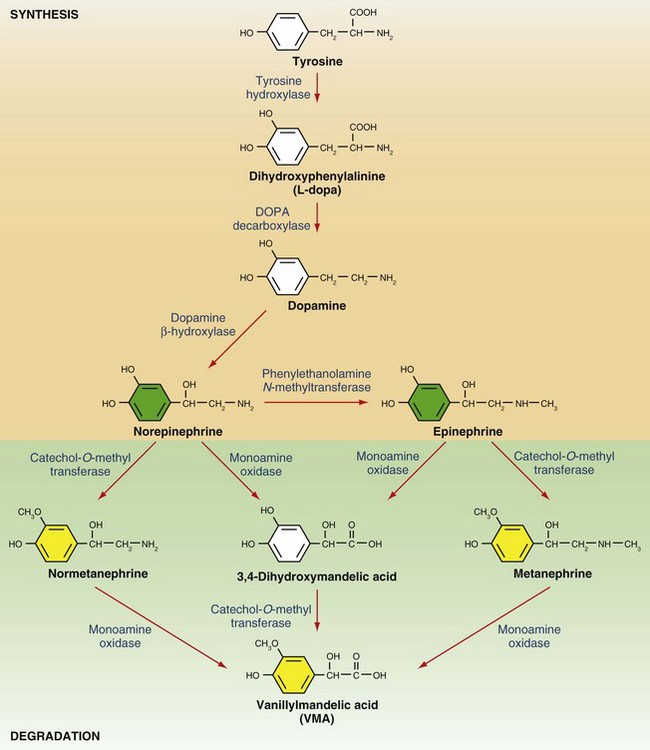
Synthesis of catecholamines in the adrenal medulla begins with the hydroxylation of tyrosine, a rate-limiting step that generates dihydroxyphenylalanine (L-dopa) in the cytosol (Fig. 41-6). Decarboxylation of L-dopa generates dopamine, which is then β-hydroxylated to form norepinephrine. Epinephrine is created by the action of PNMT, which, unlike the other enzymes involved in catecholamine synthesis, is localized to the chromaffin cells of the adrenal medulla and organ of Zuckerkandl. Sympathetic stimulation of the adrenal medulla results in the release of stored catecholamines into the circulation. Basal levels of adrenal catecholamine secretion are normally low, although large (up to 50-fold) increases in levels may be observed in response to major physiologic or psychological stressors. Target tissue responses are mediated by α- and β-adrenergic receptors. α-Adrenergic receptors display greater affinity for norepinephrine compared with epinephrine, and the opposite is true for β-adrenergic receptors. Stimulation of β1-adrenergic receptors in the myocardium results in increased heart rate and contractility. Stimulation of β2-adrenergic receptors results in smooth muscle relaxation in tissues such as the uterus, bronchi, and skeletal muscle arterioles. α1-Adrenergic receptors mediate vasoconstriction in tissues such as the skin and gastrointestinal tract. α2-Adrenergic receptors exist in presynaptic locations in the central nervous system, where they mediate attenuation of sympathetic outflow. The net effect of adrenal catecholamine release is to augment blood flow and oxygen delivery to the brain, heart, and skeletal muscle, which are essential to the fight-or-flight response, at the expense of other organ systems.
Catecholamine Clearance
Catecholamines are potent and short-acting compounds, with a plasma half-life of approximately 1 minute. Their presence in synapses and the circulation exhibits tight negative regulation by reuptake and degradation. Degradation pathways merit some discussion because they generate the metabolites commonly measured in the biochemical evaluation of pheochromocytoma (see later). Epinephrine and norepinephrine are inactivated by one or both of the enzymes monoamine oxidase (MAO) and catechol-O-methyl transferase (COMT; see Fig. 41-6). Initial methylation by COMT yields metanephrine and normetanephrine, which can be detected in plasma and urine. Their relatively stable plasma levels, which contrast with the high-amplitude fluctuations seen in plasma epinephrine and norepinephrine levels, make them attractive diagnostic markers.3 The sequential action of MAO and COMT generates the major final product, vanillylmandelic acid (VMA). Catecholamine metabolites are excreted in the urine, sometimes after sulfonation or conjugation to glucuronic acid in the liver.
Adrenal Insufficiency
Types of Adrenal Insufficiency
Primary Adrenal Insufficiency (Addison’s Disease)
Originally described in patients with tuberculous destruction of the adrenal glands, this rare disease presents with weakness and fatigue, anorexia, nausea or vomiting, weight loss, hyperpigmentation, hypotension, and electrolyte disturbances (hyponatremia and hyperkalemia). Hyperpigmentation, previously believed to be caused by elevated levels of POMC and its cleavage product alpha–melanocyte-stimulating hormone (α-MSH) is now believed to result from ACTH-induced melanogenesis.4 Hormonal insufficiency caused by intrinsic adrenal disease arises from three general mechanisms—congenital adrenal dysgenesis/hypoplasia, defective steroidogenesis, and adrenal destruction. Of these, adrenal destruction from autoimmune causes is the most common, followed by infectious adrenalitis (e.g., tuberculous, fungal, viral), adrenal replacement by metastatic tumor, and adrenal hemorrhage (Waterhouse-Friderichsen syndrome [WFS]). The latter occurs in the setting of septicemia caused by meningococcal or other organisms and is more common in pediatric and asplenic patients.
Secondary Adrenal Insufficiency
Secondary adrenal insufficiency is a relatively common disorder resulting from ACTH deficiency, often occurring in the setting of pharmacologic steroid withdrawal. Patients receiving high supraphysiologic doses of glucocorticoids (more than the equivalent of 20 mg prednisone daily; see Table 41-1) for more than 5 days and those receiving low supraphysiologic doses for more than 3 weeks are at risk for HPA axis suppression. Surgical cure of Cushing’s syndrome (see later) similarly results in glucocorticoid withdrawal. The rate of recovery from HPA axis suppression varies in accordance with the duration and severity of previous glucocorticoid excess, and the need for glucocorticoid supplementation may last several years.5 Other less common causes of secondary adrenal insufficiency include panhypopituitarism caused by neoplastic or infiltrative replacement, granulomatous disease, and pituitary hemorrhage or infarction. Pituitary infarction may occur in the setting of severe postpartum hemorrhage (Sheehan’s syndrome).
Adrenal Insufficiency in the Critically Ill
Studies have suggested that critically ill patients with sepsis or systemic inflammatory response syndrome (SIRS) may be affected by acute reversible dysfunction of the HPA axis. The incidence of the disorder is approximately 30% in critically ill patients, although this figure may be higher in those with septic shock. Whether these patients incur increased mortality because of adrenal insufficiency remains to be defined. Proposed mechanisms of reversible HPA axis dysfunction include adrenal ACTH resistance and decreased responsiveness of target tissues to glucocorticoids. Glucocorticoid supplementation in septic patients has been the topic of at least 14 randomized controlled trials. Among these studies, there appears to be an inverse relationship between survival benefit and glucocorticoid dose, with physiologic (i.e., replacement) doses yielding a median relative survival benefit of 1.33 and high supraphysiologic doses demonstrating significant harm. Although the data remains controversial, evidence has suggested that patients with vasopressor-dependent septic shock may benefit from 5- to 7-day courses of glucocorticoids in the dosage range of 400 mg/day or less of hydrocortisone or equivalent.6
Diagnosis and Treatment
Diagnosis
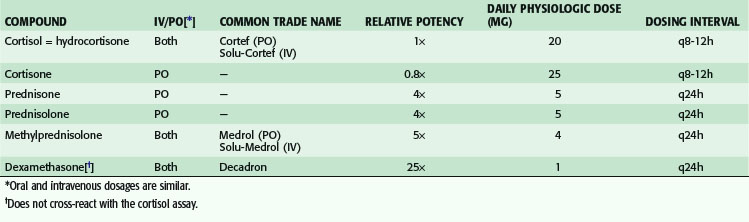
As is true for most endocrine disorders, the diagnosis of adrenal insufficiency depends on maintaining sufficient clinical suspicion for the disease. The clinical manifestations have been discussed earlier. Surgeons are most likely to encounter patients with adrenal insufficiency in the intensive care unit, trauma suite, or operating room when treating patients with steroid-dependent chronic illnesses. Routine and provocative biochemical testing is necessary to confirm the diagnosis (Fig. 41-7). The first step is to document inadequate cortisol production, which can be done by measuring morning levels of cortisol in the serum or saliva. In most patients, morning serum cortisol concentration higher than 15 µg/dL or morning salivary cortisol concentration higher than 5.8 ng/mL effectively excludes adrenal insufficiency. Patients whose levels fall below these thresholds should undergo provocative testing. A high-dose cosyntropin stimulation test is performed by administering 250 µg cosyntropin and measuring serum cortisol levels 30 to 60 minutes later. A positive test (i.e., a stimulated cortisol level lower than 18 µg/dL) is strongly suggestive of adrenal insufficiency. After the diagnosis of adrenal insufficiency has been made, a morning ACTH level is determined to differentiate between primary and secondary adrenal insufficiency.
Perioperative Steroid Administration
Recommendations concerning glucocorticoid administration during elective surgery have been based primarily on uncontrolled retrospective studies. The need for supraphysiologic doses of glucocorticoids in this setting has generally been overstated. Patients with secondary adrenal insufficiency caused by chronic glucocorticoid treatment for autoimmune or inflammatory conditions have a 1% to 2% risk of hypotensive crisis without perioperative glucocorticoid coverage. To prevent this rare but hazardous complication, chronic glucocorticoid users should, at the least, be maintained on their usual glucocorticoid dosage throughout the perioperative period. Supplementation above this level should be given in short courses according to the guidelines listed in Table 41-2.7 Patients undergoing unilateral adrenalectomy should only be given supplemental glucocorticoids if the underlying diagnosis is Cushing’s syndrome.
Table 41-2 Perioperative Glucocorticoid Regimens for Patients With Secondary Adrenal Insufficiency*
| DEGEREE OF SURGICAL STRESS | EXAMPLES | DAILY GLUCOCORTICOID DOSE |
|---|---|---|
| Minor | Procedures under local anesthesia, most outpatient procedures, inguinal hernia repair | Hydrocortisone, 25 mg or equivalent |
| Moderate | Routine abdominal, peripheral, vascular, or orthopedic surgery | Hydrocortisone, 50-75 mg or equivalent |
| Major | Resection of gastrointestinal cancer, cardiopulmonary bypass | Hydrocortisone, 100-150 mg or equivalent |
* Caused by chronic pharmacologic steroid use.
Adapted from Salem M, Tainsh RE Jr, Bromberg J, et al: Perioperative glucocorticoid coverage. A reassessment 42 years after emergence of a problem. Ann Surg 219:416–425, 1994.
Diseases of the Adrenal Cortex
Primary Hyperaldosteronism
Epidemiology and Clinical Features
Primary hyperaldosteronism, the unregulated release of excess aldosterone from one or both adrenal glands, was first described by Jerome Conn in 1954. Primary hyperaldosteronism classically presents with resistant hypertension and hypokalemia, although studies have revealed that most patients may be normokalemic, depending on the population screened. Hypokalemia is likely a manifestation of severe or late-stage disease. The prevalence of primary hyperaldosteronism has been the topic of considerable debate. It was generally believed to affect approximately 1% of hypertensives. Widespread application of the aldosterone-to-renin ratio (see later) as a screening test in certain centers has led to reports of a 10% to 40% prevalence of primary hyperaldosteronism among hypertensive patients.8 There is some consensus that these higher figures reflect strong referral bias, and that the actual prevalence in unselected hypertensive patients is likely to be 7% or less. Nonselective use of the aldosterone-to-renin ratio to identify patients with primary hyperaldosteronism is known to decrease the fraction of patients with surgically correctible disease (unilateral aldosteronoma) significantly, although the absolute number of surgically treatable cases has increased.9
Primary hyperaldosteronism is a potentially curable cause of significant cardiovascular disease. A study comparing 124 subjects with biochemically confirmed primary hyperaldosteronism with hypertensive controls matched for age and systolic blood pressure has revealed that primary hyperaldosteronism is associated with a significantly increased risk of stroke, myocardial infarction, atrial fibrillation, and left ventricular hypertrophy.10 These results add to existing evidence indicating that the adverse cardiovascular sequelae of primary hyperaldosteronism are more pronounced than those caused by blood pressure elevation alone. Successful removal of an aldosteronoma leads to regression of many of these adverse physiologic changes.
The most common causes of primary hyperaldosteronism are unilateral aldosterone-producing adenomas (aldosteronomas; Fig. 41-8) and bilateral adrenal hyperplasia (also termed idiopathic hyperaldosteronism; Table 41-3). In the past, aldosteronoma was present in more than 60% of cases, but this number has decreased substantially as nonselective screening with the aldosterone-to-renin ratio has been applied. This phenomenon may reflect increased detection of hyperplasia, which is characterized by milder biochemical abnormalities than aldosteronoma.
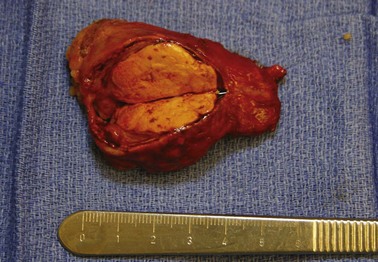
Table 41-3 Causes of Primary Hyperaldosteronism (%)*
| Screening | ||
|---|---|---|
| CAUSE | SELECTIVE | NONSELECTIVE |
| Aldosterone-producing adenoma | 60 | 30 |
| Bilateral adrenal hyperplasia (idiopathic hyperaldosteronism) | 35 | 65 |
| Aldosterone-producing adrenocortical carcinoma | <1 | <1 |
| Familial hyperaldosteronism | ||
| Type 1 (glucocorticoid-remediable aldosteronism) | <1 | <1 |
| Type 2 (non-glucocorticoid-remediable aldosteronism) | <1 | <1 |
* Rates of specific pathologies are highly dependent on the pattern of screening (selective versus nonselective).
Diagnosis and Localization
Biochemical Diagnosis
The goal of diagnostic testing is to identify and lateralize aldosteronomas. There is some consensus that biochemical screening should be performed in all patients with hypertension and unexplained hypokalemia, as well as those with hypertension sufficiently resistant to medical therapy to warrant investigation for secondary hypertension. Establishing the diagnosis of primary hyperaldosteronism begins with determining the ratio of plasma aldosterone concentration to plasma renin activity (expressed here as ng/dL divided by ng/[mL • hr]; Fig. 41-9). This test should be performed after discontinuation of interfering medications, such as spironolactone, angiotensin-converting enzyme (ACE) inhibitors, diuretics, and β-adrenergic blockers. Variable cutoff values have been used in the literature, but a value of 30 yields a sensitivity of approximately 90%.11 A subset of patients with essential hypertension will have suppressed renin levels, which may result in false elevations of the aldosterone-to-renin ratio. Thus, the inclusion of an absolute aldosterone concentration higher than 15 mg/dL increases the specificity of initial screening. Patients who test positive and are younger than 30 years should be genetically screened for glucocorticoid-remediable aldosteronism (familial hyperaldosteronism type I), especially if they have a family history of early-onset hypertension. This rare autosomal dominant condition results in abnormal regulation of aldosterone synthesis by ACTH and can be medically treated.
Localization
After the diagnosis has been confirmed, localization is performed with anatomic imaging, selective venous sampling, and sometimes functional scanning. The fact that most aldosteronomas are smaller than 15 mm in maximum dimension poses some challenges to localization. Thin-cut (3-mm) adrenal computed tomography (CT) scanning is the preferred initial localization test (Fig. 41-10).
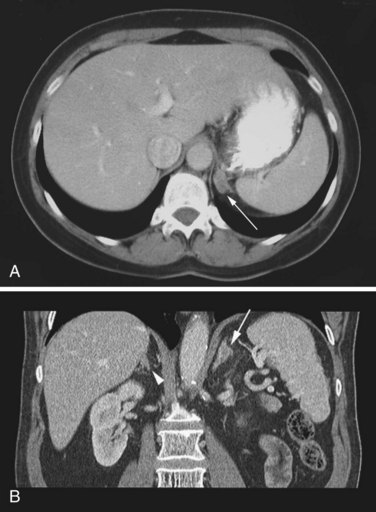
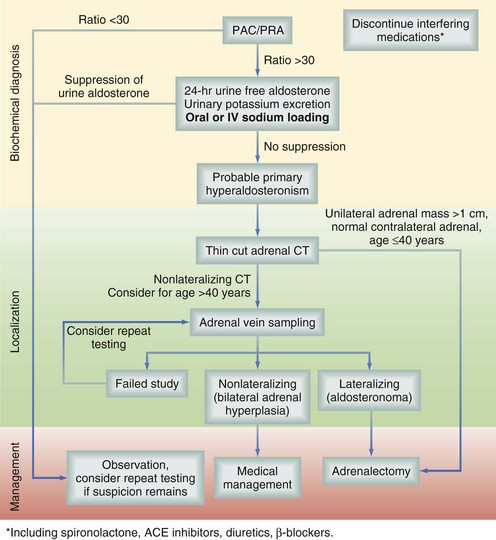
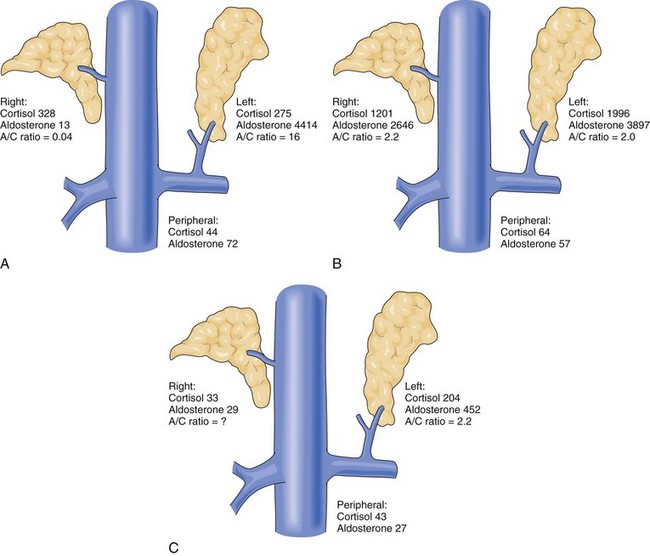
The next step in the localization algorithm is selective adrenal venous sampling (AVS). This test relies on the simultaneous measurement of cortisol and aldosterone levels in the peripheral circulation and left and right adrenal veins (Fig. 41-11). More than a fivefold elevation of the cortisol concentration in a sample relative to peripheral blood indicates successful cannulation of an adrenal vein (positive control). Lateralization is indicated by an unbalanced ratio of aldosterone to cortisol in the left and right adrenal veins, with the ratio on one side being fourfold higher than the other to identify the culprit gland. Considerable controversy exists over which patients should undergo AVS, an invasive procedure with a 90% technical success rate in experienced hands. There is consensus that AVS should be applied in all cases in which the biochemical diagnosis of primary hyperaldosteronism has been confirmed and thin-cut adrenal CT reveals no abnormalities or bilateral abnormalities. Of the remaining patients who have a unilateral mass on CT scan, a small but not insignificant fraction (2% to 10%) will represent false-positive localization and have persistent hyperaldosteronism after unilateral adrenalectomy. In these patients, the adrenal mass represents a nonfunctioning cortical adenoma and the true underlying diagnosis is a contralateral microaldosteronoma or bilateral adrenal hyperplasia, the latter of which is not surgically remediable.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


