The Adolescent and Adult with Congenital Heart Disease
Ali N. Zaidi
Curt J. Daniels
Prevalence of Adult Congenital Heart Disease (ACHD)
Most patients born with moderate or severe forms of congenital heart disease in the 1950s and earlier died before reaching adulthood, with the highest mortality during the first week of life (1). Remarkable advances in diagnostic methods, medical management, interventional techniques, congenital heart disease surgery, and perioperative care have led to historical shifts in population demographic characteristics, and adults with congenital heart defects now outnumber children by a ratio of 2:1 (2,3,4). In the United States, there are >20,000 new patients reaching adolescence each year (Fig. 67.1). With improving care, it is now expected that >90% of patients born with CHD will survive to adulthood; therefore, the already higher number of ACHD patients will continue to rise (Fig. 67.2). The prevalence of complex congenital heart disease in adults has been steadily increasing (e.g., by 85% from 1985 to 2000), unlike the relatively stable prevalence of complex congenital heart disease in children. By 2010, adults accounted for >60% of patients with complex congenital heart disease, thereby outnumbering the children with CHD (Fig. 67.3) (5). A Quebec population-based study estimated that in the year 2010 the prevalence of congenital heart disease in adults (18 years of age and older) was 6.1 per 1,000 (4). This was comparable to the results of a systematic review by Mulder et al. in the Netherlands (6). Extrapolating these statistics to the general population, it can be estimated that there are >100,000 adults with congenital heart disease in Canada, >1 million in the United States, and >1.8 million in Europe (7,8).
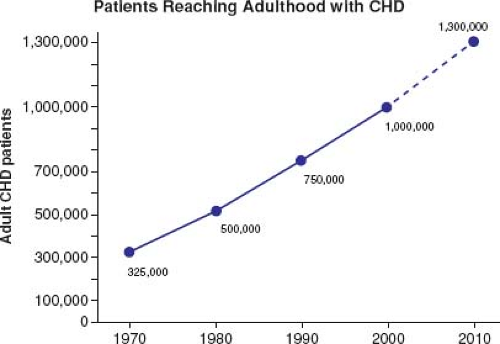 Figure 67.1 Expanding population of adults with CHD by year. |
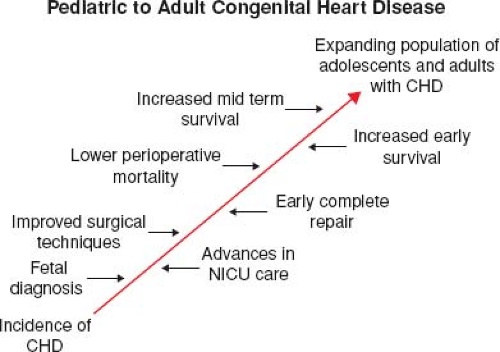 Figure 67.2 Factors leading to the expanding population of adults with congenital heart disease. |
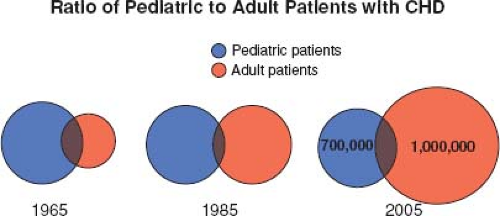 Figure 67.3 Estimated ratio of pediatric to adult patients with CHD. |
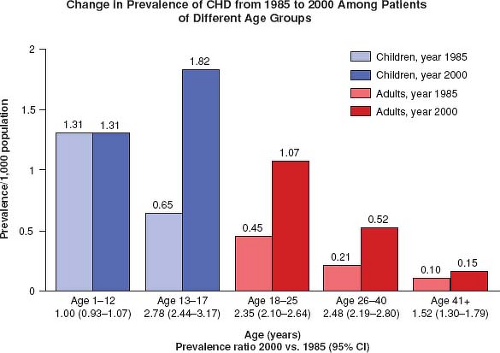 Figure 67.4 Change in prevalence of CHD from 1985 to 2000 among patients of different age groups. The highest increase in prevalence has occurred in the 13- to 17-year age group followed by the 18- to 40-year-old group. |
Marelli et al. estimated the prevalence of CHD from 1985 to 2000 for children and adults and also found a shift in the population living with CHD to those >18 years of age (Fig. 67.4) (5).
The success of pediatric cardiology and CHD surgery is tempered by the long-term complications in adult patients that may involve every subspecialty within the field of cardiology (Fig. 67.5). Common complications that contribute to the growing epidemic of adults with congenital heart disease include arrhythmias, heart failure (HF), pulmonary hypertension (PHTN), endocarditis, pregnancy-related issues, and cardiac interventions (9). By far, the most concerning of these long-term complications remains sudden cardiac death (SCD) (Fig. 67.6) (10).
An Adult Congenital Heart Disease Program
Since most congenital heart defects are not curable and require lifelong specialized care. Medical and surgical breakthroughs in the care of children born with heart defects have generated a growing population of adult survivors and spawned a new subspecialty of cardiology: adult congenital heart disease (9). Due to this rapid growth of ACHD, healthcare systems worldwide are challenged to meet the unique needs of this increasingly complex patient population, including the development of ACHD clinics and centers of excellence in order to provide comprehensive and multidisciplinary specialized care.
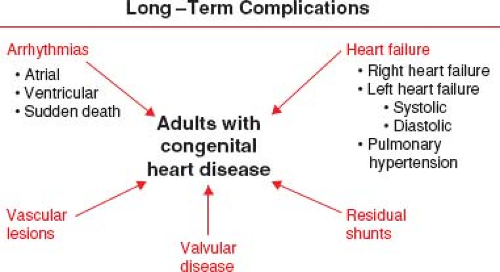 Figure 67.5 Long-term complications and residual cardiac abnormalities facing ACHD. |
Although the provision of care for children with congenital heart disease is well established in developed countries, clinical services for adults with CHD continue to be comparatively scarce (11). To attend to the progressive increase in the number of adults with congenital heart disease and the increasing complexity of their
pathologies, specialized adult congenital heart clinics have emerged in several countries, with increasing burden of activities (9).
pathologies, specialized adult congenital heart clinics have emerged in several countries, with increasing burden of activities (9).
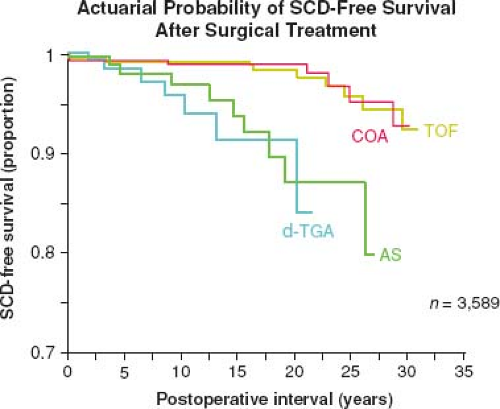 Figure 67.6 Risk of sudden cardiac death (SCD) versus years following operative repair for CHD. TOF, tetralogy of Fallot; COA, coarctation of the aorta; AS, aortic stenosis; d-TGA, d-transposition of the great arteries. |
The ACHD cardiologist who deals with these patients must therefore be familiar with congenital heart lesions in their uncomplicated state knowing appropriate testing and follow-up. Most importantly, they must also provide expert care for both natural and unnatural (surgical) consequences, and be qualified to evaluate and treat residual lesions, arrhythmias, HF and manage high-risk pregnancies in this growing population. Current management guidelines suggest that approximately half of the adult population with congenital heart disease stands to benefit from specialized care within adult congenital heart centers. Many patients with simple lesions (e.g., atrial septal defects [ASDs], ventricular septal defects [VSDs], patent ductus arteriosus [PDA], and mild pulmonary stenosis) who have undergone total corrective surgery will have few, if any, hemodynamic residua and require infrequent evaluation and treatment. Patients with more complex lesions, or complications that stem from less complex lesions, such as residual shunts, endocarditis, valvular disease, ventricular dysfunction, aortopathies and arrhythmias require more frequent evaluation, medical treatment, and consideration for further surgical- or catheter-based interventions.
As we continue to learn about surgically altered congenital heart diseases, some “routine” patients will have previously unrecognized problems. For other adults, surgical approaches of the past and their long-term complications (e.g., TGA s/p Mustard or Senning Atrial Repair) will eventually become obsolete and replaced by new complications of present day surgical repair. Such specialized care is generally recommended for the initial assessment of adults with known or suspected congenital heart disease, follow-up of patients with moderate and severe lesions, cardiac surgical and nonsurgical interventions, and risk assessment and support for pregnancy and noncardiac surgery (12,13). It is now well known that centralized care within regional ACHD centers should be provided by a multidisciplinary team comprised of cardiologists with expertise in adult congenital heart disease, including imaging, arrhythmia management, interventional cardiology, and high-risk obstetrics, congenital heart surgeons, perioperative care, and psychosocial support (12,13). With adult congenital heart disease clinics/centers seeing more patients because of improved survival, there continues to be a need for more specialized ACHD cardiologists and centers across the United States. At one of the largest centers for ACHD in the nation, the Columbus Ohio Adult Congenital Heart Disease Program (COACH) at The Nationwide Children’s Hospital and The Ohio State University has experienced an exponential rise in all areas of clinical care, attesting to the expanding population and the need for ongoing appropriate care for this population (Fig. 67.7).
However, an adequate ratio of specialized adult congenital centers is no guarantee for optimal care. A larger issue that plagues the field of congenital heart disease is the relatively small proportion of qualifying patients who actually receive specialized adult-oriented care as they transition from pediatric cardiology into the realm of adult medicine. There are several factors associated with “gaps in care” and impediments to long-term follow-up. Common barriers to transfer from pediatric- to adult-oriented health care include
inadequate knowledge regarding the need for such care, a dearth of accessible competent adult care providers, and emotional attachment of patients and families to their pediatric caregivers and vice versa (14). Rather disturbing is the discovery that the same patient who was evaluated annually by a pediatric cardiologist, saw a cardiologist only every 10 years after reaching the age of 21 (15). This indicates that a better and more accessible system must be provided to these patients.
inadequate knowledge regarding the need for such care, a dearth of accessible competent adult care providers, and emotional attachment of patients and families to their pediatric caregivers and vice versa (14). Rather disturbing is the discovery that the same patient who was evaluated annually by a pediatric cardiologist, saw a cardiologist only every 10 years after reaching the age of 21 (15). This indicates that a better and more accessible system must be provided to these patients.
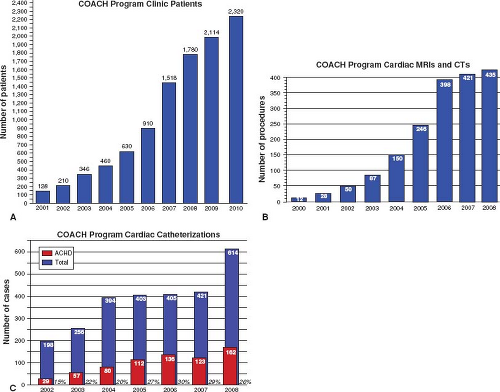 Figure 67.7 A: ACHD clinic, (B) cardiac MRI/CT, and (C) cardiac catheterization volume since 2000 at The Columbus Ohio Adult Congenital Heart Disease (COACH) Program at the Nationwide Children’s Hospital and The Ohio State University. |
Because of lack of training and interest, some pediatric cardiologists, who are the best trained to understand most of the defects encountered in this population, do not remain involved in the long-term care of these patients as they get older. Beyond that, many internal medicine cardiologists have had minimal exposure to congenital heart disease during their training or experience, yet are expected to manage even the most complex of these patients.
In specialized ACHD clinics/centers, the staffing, ambience, and style of evaluation are different from the setting of newborns and toddlers with CHD. The clinic should be held separately from that for younger patients, and should be geared toward the age of the patient population. Depending on the patient’s age and maturity, parents may or may not accompany the patient to the office visit. Some patients will come with their spouses, and some will even bring their own children. Since many are young adults, unless the individual’s development level prohibits understanding, the practitioner should deal with the patient privately, as well as with those who accompany the patient. The adolescent and adult should understand that the cardiologist is willing to discuss certain issues in privacy and that these discussions are confidential. By this age, these patients should share in or primarily decide their courses of therapy and behaviors. “Weaning” the young adult from parental decision making is one of the major challenges facing patients as they reach adulthood. The ongoing management of this complex patient population can often be challenging in the subset of patients with ACHD who have multiple physical and/or mental disabilities. It is not uncommon for adults with CHD to be living with their parents and to have complex psychosocial needs. Continuity of care may also be compromised by noncompliance in the presence of minimal or no symptoms or with the denial phase, which is common during adolescence. Finally, patients may be lost to follow-up when they relocate geographically for education or work-related reasons (16).
Staffing adolescent and adult cardiac clinic varies from institution to institution. Such a team may include experienced pediatric cardiologists, internal medicine cardiologists, cardiac surgeons experienced in dealing with congenital heart defects, nurse clinicians, psychologists, and social workers. The milieu should include trainees from both pediatric and internal medicine disciplines so that a more coordinated effort can be secured for the future. The reality is that there are very few pediatric cardiologists compared with the number of internal medicine cardiologists, and that the transition of many of these patients to adult cardiologists is inevitable. Those providing care must be trained and have the ability to care for this unique population. These ACHD centers should have consultative access to obstetricians and gynecologists, psychiatrists, endocrinologists, nephrologists, hematologists, rheumatologists, pulmonologists, anesthesiologists, and pathologists, all with an understanding of congenital heart disease and its impact on this population.
In addition to assisting their parents through a sophisticated understanding of their heart problems, the practitioner in the ACHD clinic is expected to help the patient manage lifestyle issues. This includes sexuality (including contraception, pregnancy, and evaluation of offspring), education and employability, insurability, and exercise and athletics (16). Current management guidelines suggest that approximately half of the adult population with congenital heart disease stands to benefit from specialized care within adult congenital heart centers. Such care is generally recommended for the initial assessment of adults with known or suspected congenital heart disease, follow-up of patients with moderate and severe lesions, cardiac surgical and nonsurgical interventions, and risk assessment and support for pregnancy and noncardiac surgery (12,13).
Although the recommended number of specialized centers has been achieved in Canada (i.e., 17 centers are registered within the CACH network), most countries in the developed world fall short of this target. For example, in the United States, 108 self-declared adult congenital heart centers with variable volumes of activity and availability of resources are listed on the American Congenital Heart Association (www.achaheart.org). Despite the growing number of such ACHD centers, only a fraction of the ACHD patients are seen in such centers. A Canadian study found that <25% of adults with congenital heart disease are followed by specialized centers and that waiting times to access services exceed published recommendations (17).
Training in Adult Congenital Heart Disease
An important element in optimizing outcomes is the development of training programs to meet workforce requirements and provide qualified, consistent, and comprehensive care. Exposure of adult cardiology trainees to adult congenital heart disease didactic and clinical experience varies widely, reflecting the fact that few programs offer dedicated advanced training in this section of cardiology (18). Prior surveys have suggested that only 25% of cardiologists who care for adults with congenital heart disease have received formal training in this discipline (17). In fact, internal medicine cardiology training only requires 6 hours of congenital heart disease lectures to be board eligible for internal medicine cardiovascular boards.
There are only a few dedicated ACHD experts currently, due to the need for both congenital heart disease experience and adult medicine training. This lack of dedicated ACHD cardiologists has led to an important element in optimizing outcomes in the development of training programs to meet workforce requirements and provide qualified, consistent, and comprehensive care. The United States is the first country to officially recognize the legitimacy of adult congenital heart disease as a separate cardiology subspecialty. In September 2012, the American Board of Medical Subspecialties approved a specialized certification examination via the American Board of Internal Medicine. The first examination is planned for 2015. Pediatric or adult cardiologists require a minimum 2-year fellowship within recognized adult congenital training centers to qualify (9). The ESC, with its working group on grown-up congenital heart disease, is poised to follow suit (13). Building on the work of the Canadian Adult Congenital Heart Network established in 1992, the Canadian Cardiac Society (CCS) convened a panel of international experts for an ACHD consensus conference in 1996. Foremost among the proposals arising from this forum was a recommendation that all ACHD patients be referred to specialized ACHD centers for ongoing care (19). Clinical guidelines to this effect were presented at the 1996 CCS conference, published in 1998 (12), and were followed by similar proposals emerging from the 32nd Bethesda Conference in 2001 (20). Despite these recommendations and more than a decade later, less than one-third of eligible patients actively receive specialized ACHD care in Canada, where health insurance is universal (17). Gurvitz et al. showed that in the United States, this proportion is expected to be significantly less, and care gaps and dispersion have been documented (14,21).
In a large population-based study, Marelli et al. showed that a significant rise in referral to specialized ACHD centers was observed concurrent with national consensus guidelines. Importantly, a significant reduction in mortality was associated with referral to an ACHD center and was most pronounced in patients with severe forms of CHD. They also showed that specialized ACHD care was independently associated with reduced ACHD patient
mortality when tested in both case-control and cohort studies. This observation was robust despite a higher incidence of severe CHD in those treated in ACHD referral centers. Advances in diagnosis and surgical treatment of CHD are expected to have impacted survival, and, although these advances were potentially available to both ACHD referral and nonreferral centers, the increased accessibility to expertise and application of tertiary and quaternary care by caregivers specialized in ACHD can explain the protective effect of ACHD referral centers (22). This, in fact, constitutes the rationale for the recommendations that all ACHD patients be assessed in a specialized ACHD center at least once, and that patients with moderate or complex CHD be followed periodically or consistently in specialized ACHD centers (12,13).
mortality when tested in both case-control and cohort studies. This observation was robust despite a higher incidence of severe CHD in those treated in ACHD referral centers. Advances in diagnosis and surgical treatment of CHD are expected to have impacted survival, and, although these advances were potentially available to both ACHD referral and nonreferral centers, the increased accessibility to expertise and application of tertiary and quaternary care by caregivers specialized in ACHD can explain the protective effect of ACHD referral centers (22). This, in fact, constitutes the rationale for the recommendations that all ACHD patients be assessed in a specialized ACHD center at least once, and that patients with moderate or complex CHD be followed periodically or consistently in specialized ACHD centers (12,13).
Despite surgical repair, most forms of congenital heart disease are associated with sequelae and long-term complications which engender a high level of healthcare resource utilization (23). Rates of emergency room visits, hospital admissions, and cardiac interventions are severalfold greater in ACHD than in the general population. Models of health-related costs and outcomes in adults with congenital heart disease provide economic justification for expensive interventions on the basis of striking benefits on quality-adjusted life years (24).
In 2005, more than three-quarters of ACHD patients did not have an outpatient cardiology review (4). The observation that less than one in three ACHD patients actively receives care at a specialized center suggests that the need for delivery of appropriate health care to ACHD patients remains largely unmet (17).
Specialized ACHD centers should provide a range of services specifically geared toward meeting the needs of this patient population. Physician expertise is key to delivering quality ACHD care such that ACHD subspecialty training programs have emerged to address the shortage of manpower in this emerging field of cardiology (25).
The following section will discuss the long-term sequelae of the most common congenital heart disease lesions presenting in adulthood with or without prior repair, and the long-term complications, diagnostic workup and therapeutic options that are deemed necessary.
Bicuspid Aortic Valve
The congenitally bicuspid aortic valve (BAV) is the most common congenital malformation occurring in approximately 2% of the general population (26). It is the most common cause of isolated valvular aortic stenosis in adults, with a male to female ratio of 3:1 (27). The BAV may be functionally normal with no significant pressure gradient across the valve, with no more than trace valvular aortic regurgitation (AR). However, thickening and focal calcification of the bicuspid valve can be detected as early as the second decade of life (28). The most common presenting sign or symptom in a young adult is often the detection of a systolic ejection murmur and usually an ejection click. However, depending on the severity of valvular disease, patients with aortic stenosis or aortic insufficiency may present with exercise intolerance, dyspnea on exertion, or atypical chest pain with the peak incidence of symptoms (angina, syncope, or HF) developing with advancing years.
Those without significant aortic stenosis (usually defined as a gradient less than 25 mm Hg) and less than mild aortic insufficiency only require regular follow-up. However, over time, the lesion often progresses due to fibrocalcific stenosis with almost 75% of patients requiring eventual surgery (29). The joint study on the Natural History of Congenital Heart Defects followed 473 patients with aortic valve disease a mean of 20 years. Only 20% of those with an initial peak to peak gradient <25 mm Hg at catheterization had a subsequent intervention. However, in those with a gradient >50 mm Hg, arrhythmias, sudden death, endocarditis, syncope, and angina occurred at a rate of 1.2% per year (15).
The risk factors that lead to the development of aortic stenosis in patients with BAVs are variable and have been poorly defined, often thought to be related to valvular characteristics. Beppu et al. performed an echocardiographic study evaluating 75 patients (15 to 76 years of age) with a BAV and found that aortic valve sclerosis began around the second decade of life and aortic calcification started in the fourth decade. Aortic valve pressure gradient increased approximately 18 mm Hg each decade, concomitant with valve sclerosis. Patients with anteroposteriorly (as opposed to right-left), and eccentric (vs. symmetric) valve leaflets had a faster rate of progression with an aortic valve pressure gradient increasing an average of 27 mm Hg per decade (30).
Patients with BAVs should be monitored for progressive aortic valve dysfunction (stenosis and/or regurgitation) as well as for aortic dilation with risk of aneurysm formation and aortic dissection. Echocardiography should be performed at intervals, based upon the lesion requiring greatest frequency of surveillance among aortic stenosis, AR, and dilation of the aorta. The rate of aortic growth in BAV patients is variable, ranging from 0.2 to 0.9 mm/year, depending upon patient characteristics (31). Development of significant aortic stenosis is much more common than development of significant AR.
Based on the 2014 American Heart Association/American College of Cardiology (AHA/ACC) valve guidelines and the 2008 ACC/AHA adult congenital heart disease guidelines and are consistent with the 2011 appropriate use criteria for echocardiography, the following recommendations are suggested for surveillance for aortic stenosis and aortic dilation in patients with BAV (12,32,33).
For asymptomatic adolescents and young adults (<30 years of age) with BAV:
With aortic valve mean Doppler gradient >30 mm Hg or peak instantaneous gradient >50 mm Hg, yearly Doppler echocardiography and a yearly electrocardiogram (ECG).
With aortic valve mean Doppler gradient <30 mm Hg or peak instantaneous gradient <50 mm Hg, we suggest Doppler echocardiography and an ECG every 2 years.
For older adults (>30 years of age) with aortic stenosis:
With severe aortic stenosis (defined as peak jet velocity ≥4 m/s, mean gradient >40 mm Hg, valve area <1.0 cm2), Doppler echocardiography every 6 to 12 months.
With moderate aortic stenosis (defined as peak jet velocity 3 to 3.9 m/s, mean gradient 25 to 40 mm Hg, valve area 1.0 to 1.5 cm2), Doppler echocardiography every 1 to 2 years.
With mild aortic stenosis (defined as peak jet velocity 2.0 to 2.9 m/s, mean gradient <25 mm Hg, or valve area >1.5 cm2), Doppler echocardiography every 3 to 5 years.
For adults of all ages with BAV and an aortic diameter >4.0 cm:
Serial evaluation of the size and morphology of the aortic sinuses and ascending aorta by echocardiography, cardiovascular magnetic resonance, or computed tomography (CT) angiography is recommended, with the examination interval determined by the degree and rate of progression of aortic dilation and by family history.
If the aortic root or ascending aorta diameter is >4.5 cm, or if there is a rapid rate of change in aortic diameter or a family history of aortic dissection, reassessment of the aortic root and ascending aorta size should be performed at least yearly.
First-degree relatives of patients with BAV should be evaluated for the presence of BAV and thoracic aortic disease. Aortic valve disease surveillance is generally performed by transthoracic echocardiography (TTE). Aortic root and ascending aorta size are measured by two-dimensional TTE using the maximum diameter perpendicular to aortic flow. Although the 2010 ACC/AHA thoracic aortic disease guideline recommends using the internal ascending aortic diameter (34,35), echocardiographic guidelines recommend measuring the aortic annulus at the hinge points of the
aortic valve leaflets using the inner edge to inner edge. If the aortic root and ascending aorta above the sinotubular junction cannot be adequately visualized by echocardiography, noncontrast magnetic resonance imaging (MRI) or contrast CT is recommended. On MRI or contrast CT images, the external diameter is used. In patients requiring serial imaging, MRI may be preferred to avoid repeated radiation exposure. In patients with impaired renal function or a history of shellfish allergy, the risk of iodinated intravenous contrast for contrast CT should be considered.
aortic valve leaflets using the inner edge to inner edge. If the aortic root and ascending aorta above the sinotubular junction cannot be adequately visualized by echocardiography, noncontrast magnetic resonance imaging (MRI) or contrast CT is recommended. On MRI or contrast CT images, the external diameter is used. In patients requiring serial imaging, MRI may be preferred to avoid repeated radiation exposure. In patients with impaired renal function or a history of shellfish allergy, the risk of iodinated intravenous contrast for contrast CT should be considered.
The role of medical therapy in patients with BAV is limited since there is scarce evidence of efficacy (32). No definitive medical therapy has been found to alter the natural history of BAV disease or aortic stenosis.
Treatment modalities for patients with BAV and aortic stenosis have been defined. While percutaneous aortic valvuloplasty is not recommended in the setting of calcific aortic stenosis in older adults, there is a role for valvuloplasty in some adolescents and younger adults with aortic stenosis (most commonly due to bicuspid commissural fusion) without significant valve calcification or regurgitation. It should be considered in a selected population with significant aortic stenosis—usually defined as a peak gradient ≥60 mm Hg or ≥50 mm Hg in a symptomatic patient. These are the following recommendations adopted from the 2008 ACC/AHA adult congenital heart disease guidelines (12).
Aortic balloon valvuloplasty is recommended for adolescents or young adults and others with no AR and without significant aortic valve calcification with either of the following:
Symptoms (angina, syncope, and/or dyspnea on exertion) and a peak-to-peak gradient at catheterization greater than 50 mm Hg, OR
ST or T wave abnormalities in the left precordial leads on electrocardiogram (ECG) at rest or with exercise and a peak-to-peak gradient at catheterization greater than 60 mm Hg and no symptoms.
For asymptomatic adolescents or young adults with aortic stenosis and a peak-to-peak gradient on catheterization greater than 50 mm Hg if the patient is planning to play competitive sports or become pregnant.
Aortic balloon valvuloplasty should not be done in asymptomatic adolescents or young adults with a peak-to-peak gradient less than 40 mm Hg without symptoms or ECG changes.
Aortic balloon valvuloplasty should not be done in older adults with calcified valves as an alternative to aortic valve replacement.
In a large collaborative registry involving 606 patients, the peak-to-peak gradient was reduced by a mean of 60% after balloon valvuloplasty (36). However, this procedure should be considered palliative and patients require serial follow-up (37).
Pulmonary autograft aortic valve replacement (Ross procedure) has a role and for some is the surgical procedure of choice, especially in the adolescent and young adult with significant aortic valve disease. With a successful operation, long-term anticoagulation is not indicated and therefore the patient may not need to be restricted from most activities. The long-term follow-up of this population is promising but particular attention must include assessment of the neoaortic valve, the neoaorta and also the new pulmonary homograft, as it may progressively stenose (38,39). Mid- to long-term results of the Ross procedure have shown excellent results, however with longer follow-up patients can develop neoaortic valvular regurgitation and dilation of the neoaortic root (40,41). Patients with neo-AR and/or neoaortic root dilation need routine follow-up with serial imaging using echocardiography, CMR, or computed tomography. Whether a patient following the Ross procedure is safe to compete in contact or highly competitive sports is yet to be determined.
Although the patient with BAV is at risk for endocarditis, antibiotic prophylaxis is not recommended for isolated BAV. Good dental care and oral health practices are recommended for patients with BAV (42,43).
A BAV is frequently associated with other congenital cardiovascular defects, including coarctation of the aorta, supravalvular aortic stenosis, subvalvular aortic stenosis, VSD, and sinus of Valsalva aneurysm. The combination of BAV and coarctation of the aorta is associated with high risk of aortic complications. Less common and often previously treated lesions include ASDs or VSDs, and PDA.
Some studies provide support to the theory that the BAV is part a single developmental anomaly affecting the aortic root that eventually leads to aortic root dilation. Two lines of evidence support this theory. First of all, autopsy studies have demonstrated a 5 to 10 times increase in the incidence of aortic dissection compared to patients with tri-leaflet aortic valves. This occurred without aortic stenosis, aortic coarctation, or hypertension (41). Secondly, Warren et al. in children and Hahn et al. in adults showed that the aortic root was enlarged in patients with a BAV without aortic stenosis as compared to age and sex-matched controls (43,44). These associations have led to the theory that congenital abnormalities of the aortic valve and the aorta may reflect a common developmental defect. It is also postulated that increased risk of aortic disease in patients with BAVs (including those that function normally) is mediated by coexisting defects in the aortic media by fragmentation of elastic, loss of smooth muscle cells, and increase in collagen (45,46,47).
Therefore, those with a BAV but without significant aortic valve stenosis or regurgitation require routine assessment for aortic root enlargement. Echocardiography may be utilized to screen and follow the aortic root, but may not provide adequate imaging beyond the first few centimeters above the sinuses of Valsalva, therefore potentially missing significant enlargement in the distal ascending aorta. CMR as a screening tool is often necessary to evaluate the entire aortic root from the valve annulus to the take-off of the great vessels (Fig. 67.8). According to current guidelines, serial evaluation of a patient with a BAV and dilated ascending aorta (>4 cm) with echocardiography, CMR or MRI is recommended a yearly basis. First-degree relatives of all patients with a BAV should be screened with echocardiography (47).
The timing of prophylactic aortic root or ascending aortic replacement in the setting of BAV disease is complex. Various major society guidelines recommend differing aortic diameter thresholds (generally ranging from 5.0 to 5.5 cm) for patients with BAV. These recommendations are largely based on consensus expert opinion since only limited observational data are available.
Surgery is recommended to repair or replace the ascending aorta in patients with BAV when the aortic root or ascending aorta diameter is >5.5 cm, unless other risk factors for aortic complications are present. This recommendation is consistent with the 2014 AHA/ACC valve guideline (32). In contrast, the 2010 thoracic aortic disease guidelines recommended surgery to repair or replace the ascending aorta in patients with BAV when the ascending aorta diameter is >5.0 cm (or if the growth rate is >5 mm/year) (34). Practice guidelines suggest that beta-blockers are reasonable for nonoperative candidates with dilated (>4 cm) aortas and BAV (in the absence of significant AR), but this recommendation is based on consensus opinion rather than clinical trials (34,48).
The 2014 European Society of Cardiology (ESC) aortic diseases guidelines and 2012 ESC valve guidelines also recommend aortic surgery at an aortic diameter threshold of ≥5.5 cm or if the rate of increase in diameter is 0.5 cm per year or more, or if patients are undergoing AVR due to severe AS or AR with the diameter of the aortic root or ascending aorta being greater than 4.5 cm (49). Patients with aortic dissection involving the ascending aorta or symptoms suggestive of expansion of a thoracic aneurysm should be evaluated for prompt surgical or endovascular intervention.
Patients with a BAV undergoing isolated aortic valve replacement should continue to undergo surveillance of the aortic root and ascending aorta after surgery. Borger et al. found the 15-year freedom from aortic root complications (replacement, dissection) following aortic valve replacement was 86%, 81%, and 43% for
aortic diameters of <40 mm, 41 to 44 mm, and >45 mm, respectively at the time of aortic valve replacement (48).
aortic diameters of <40 mm, 41 to 44 mm, and >45 mm, respectively at the time of aortic valve replacement (48).
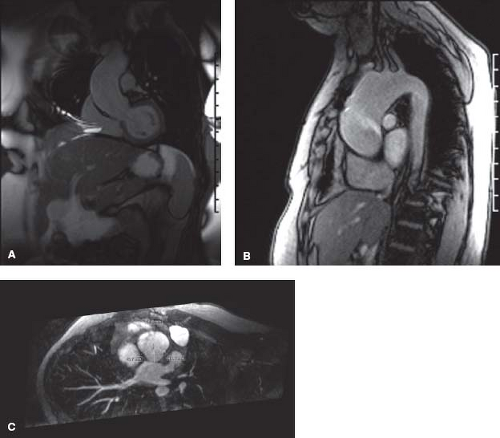 Figure 67.8 A: A 26-year-old patient with BAV with no AS/AI, CMR demonstrates aortic root enlargement measuring 50.3 cm at the widest diameter mid ascending aorta. B: A 32-year-old patient with BAV, mild AS, and large ascending aortic aneurysm measuring 5.2 cm with the widest diameter just proximal to the take-off of the right innominate artery. Echocardiography maximal diameter just above sinuses of Valsalva was 4.0 cm therefore underestimating the maximal diameter of the ascending aorta. C: Enface MRA showing dilated aortic root in 32-year-old patient with BAV. (C courtesy of Kan Hor, MD, Nationwide Children’s Hospital, Columbus, Ohio.) |
Aortic Valve Regurgitation
Aortic valve regurgitation is usually a manifestation of a congenitally abnormal aortic valve, subaortic obstruction damaging the aortic valve, aortic cusp prolapse through a supulmonic VSD, or aortic root dilation from connective tissue disorder. Pure AR is more commonly seen in young patients, and some degree of aortic stenosis coexists in older patients. BAV is considered to be the most common cause of primary AR in the developed world (50).
For most, chronic aortic valve regurgitation is well tolerated for a number of years, with slow progression but can ultimately lead to left ventricular dilation and dysfunction. Such patients are often asymptomatic for decades, until they have symptoms suggestive of left HF (dyspnea with exertion, angina, or syncope). The volume load that affects the left ventricle leads to a series of compensatory myocardial remodeling that cause left ventricular dilation. Often these changes can be reversed using appropriate pharmacologic therapy, however patients with significant aortic valve regurgitation and dilated left ventricles ultimately may require aortic valve replacement. Patients with severe AR can be treated with vasodilator therapies to alleviate the volume load on the LV (when AVR is not recommended), or to improve the hemodynamic profile and provide symptomatic relief for patients with severe HF and severe LV dysfunction (47).
The clinical examination, exercise tolerance, and LV size and function are monitored closely in patients with chronic AR who are not yet candidates for surgery. In the absence of symptoms, management decisions are largely based upon serial measurements of LV size and function. Annual echocardiographic studies are indicated in all patients with clinically significant AR. Less frequent studies may be indicated if the LV size and function remain stable over several years of follow-up.
Based on the 2014 AHA/ACC valve guidelines, the 2008 ACC/AHA adult congenital heart disease guidelines and are consistent with the 2011 appropriate use criteria for echocardiography, the following recommendations are suggested for surveillance for AR with BAV:
With severe AR, Doppler echocardiography every 6 to 12 months (more frequently if the left ventricle is dilating).
With moderate AR, Doppler echocardiography every 1 to 2 years.
Randomized trials have produced conflicting findings as to whether there is or is not a benefit from vasodilator therapy (ACE inhibitor or nifedipine) among asymptomatic patients with severe AR, LV dilation, and normal LV systolic function. In comparison, there is no evidence supporting vasodilator therapy in asymptomatic patients with severe AR without LV dilation or those with mild-to-moderate AR.
Aortic valve replacement or repair is indicated in symptomatic patients with chronic AR. The appropriate timing of aortic valve replacement for aortic valve regurgitation is partially based on the development of symptoms. Symptomatic adults (with angina, syncope, or dyspnea with exertion) with chronic severe AR should undergo AVR. Aortic valve replacement or repair is indicated in asymptomatic patients with chronic severe AR who have reliable evidence of LV systolic dysfunction (left ventricular ejection fraction [LVEF] <50%). In addition, aortic valve replacement is suggested in asymptomatic patients if LVEF is >50% and LV end-systolic dimension >55 mm, or LV end-diastolic dimension >75 mm.
Surgery should be considered in asymptomatic patients who show progressive LV enlargement, particularly if the end-diastolic dimension exceeds 70 mm, the systolic dimension approaches 50 mm, or the ejection fraction (EF) approaches 50%. AVR is reasonable in patients with moderate AR (stage B) who are undergoing other cardiac surgery (32).
It should be highlighted that the presence of LV systolic dysfunction (LVEF <0.5) indicates that the patient may be entering a decompensated stage and such patients should not only undergo intense medical treatment including the use of vasodilators and diuretics to eliminate the congestive state but should undergo a thorough evaluation for AVR. New markers of severity of AR and its resultant LV volume overload are under investigation. These include measures of regurgitant fraction, regurgitant volume, effective regurgitant orifice area, LV volume assessment with three-dimensional echocardiography, noninvasive measures of LV end-systolic stress and systolic and diastolic strain rates, and biomarkers such as brain natriuretic peptide.
Subaortic Stenosis
Subvalvar AS encompasses a variety of lesions which can occur alone or in combination. These include a thin membrane (the most common lesion), thick fibromuscular ridge, diffuse tunnel-like obstruction, abnormal mitral valve attachments, and occasionally, accessory endocardial cushion tissue.
In the majority of patients, obstruction is caused by a discrete membrane or a thick fibromuscular ridge that is attached to the ventricular septum or completely encircles the left ventricular outflow tract (LVOT). Diffuse, “tunnel-like” narrowing of the LVOT is rare and is characterized by marked myocardial hypertrophy and, often, aortic annular hypoplasia.
Patients with mild-to-moderate obstruction often remain asymptomatic for several years and are often not identified until later in life. It has been proposed that subvalvar AS is due to progressive LVOT thickening and scarring due to turbulence caused by an underlying abnormality in LVOT architecture. Patients with mild or moderate obstruction are typically asymptomatic. The lesion is often uncovered during evaluation of other associated cardiac defects. When uncomplicated, it is generally identified when echocardiography is performed for evaluation of a murmur. More than one-half of the affected patients have a harsh systolic ejection murmur heard best at the mid-left sternal border. Many patients also have associated AR. Significant subaortic obstruction is associated with left ventricular hypertrophy, and often with aortic valvular regurgitation leading to operative repair.
In contrast to valvar AS, subvalvar AS does not respond to balloon dilation. Definitive therapy consists of surgical correction using simple membrane removal, to extensive ring resection with or without myomectomy or the Konno procedure. Because of the high rate of recurrence, the timing of surgery, especially in the first decade of life, is controversial. Recommendations range from early operation to longer periods of observation, varying with patient characteristics (52). The appearance of significant AR is considered an indication for surgery because it is an acquired lesion, although the extent rarely progresses to or beyond moderate in young children.
The recurrence risk in the adult patient following initial surgical resection had previously been thought to be low, based upon limited data from small, single-center series. However, in a large (313 patients) multicenter study with a median duration of follow-up of 12.9 years, 26% of patients required reoperation during the follow-up period of the study. In this cohort, nearly all patients were noted to have adequate surgical relief (median 76 mm Hg preoperatively to 15 mm Hg postoperatively) and there was an overall increase in the gradient of 1.3 mm Hg per year. Mild AR was present in most patients (68%), but generally did not progress over time. Predictors for reoperation included female sex (hazard ratio [HR] 1.5) and progression of LVOT gradient (HR 1.5) (53).
Patients with discrete membrane or fibromuscular ridge have usually undergone surgery by adulthood, however, these lesions have a tendency for re-growth and concomitant aortic valve disease (54). A large retrospective study of 75 patients postsurgical resection found in a recurrence rate of 16% at 5 years and 30% at 10 years. Patients with a higher preoperative gradient (>40 mm Hg), higher postoperative gradient (>10 mm Hg), and younger age at surgery were predictors for recurrence (55). Additionally, the need for aortic valve repair and the progressive aortic insufficiency occurred less often in those with a lower preoperative gradient. This finding has led some to recommend early repair of fixed subaortic obstruction prior to the development of high gradient or aortic valve disease (55).
Lupinetti et al. were able to demonstrate a significantly lower recurrence of subaortic obstruction requiring reoperation when membrane excision was combined with myectomy as opposed to membrane excision alone (4% vs. 25% over a mean of 4.5 and 5.2 years, respectively). However, there was a slightly higher incidence of postoperative heart block in the myectomy group (56).
An older child with a gradient <30 mm Hg may be followed medically until there is significant progression of the obstruction documented by echocardiography or catheterization, or definite progression of AR to at least a mild degree. Unoperated adults with stable gradients below 50 mm Hg and without significant left ventricular (LV) hypertrophy must also be followed closely, since some of these patients will eventually require surgery.
“Prevention” of AR alone is not a criterion for surgery, although, as noted above, the appearance and worsening of regurgitation is frequently used as a criterion for surgery. In the 2007 AHA guidelines, antibiotic prophylaxis to prevent bacterial endocarditis is no longer recommended in patients with subvalvar AS (42).
Coarctation of the Aorta
Coarctation of the aorta is a narrowing of the descending aorta, which is typically located at the insertion of the ductus arteriosus just distal to the left subclavian artery. Coarctation of the aorta is usually a postoperative concern in adults. Previously undiagnosed adults with native coarctation are rare. Most adult patients are asymptomatic, unless they have severe hypertension leading to headaches, epistaxis, HF and/or aortic dissection. Unfortunately despite adequate childhood surgery, patients are at risk for several concerning long-term complications which include: systemic hypertension, recoarctation, aortic aneurysm/dissection, or SCD.
All patients with coarctation (repaired or not) should be monitored with lifelong congenital cardiology follow-up and imaging because long-term survival is reduced compared with normative populations and there is potential need for reintervention. A long-term follow-up study of patients repaired in childhood or adolescence demonstrated a significantly reduced long-term survival—mean age
of death being 38 years (57). Patients died from, in decreasing order, coronary artery disease, congestive HF, sudden death, cerebral vascular accidents, and ruptured aortic aneurysms. Silka et al. found the risk of SCD following COA repair to be 25 times greater than expected 20 years from repair (10). The largest single-center series describing long-term outcome included 819 patients (mean age at repair 17.2 ± 13.6 years) who underwent isolated operative repair of coarctation at Mayo Clinic between 1946 and 2005. The survival rates were 93%, 86%, and 74% at 10, 20, and 30 years after primary repair, respectively (58). In a previous report from this center, the most common cause of late death was coronary artery disease, followed by sudden death, HF, cerebrovascular accident, and ruptured aortic aneurysm (57).
of death being 38 years (57). Patients died from, in decreasing order, coronary artery disease, congestive HF, sudden death, cerebral vascular accidents, and ruptured aortic aneurysms. Silka et al. found the risk of SCD following COA repair to be 25 times greater than expected 20 years from repair (10). The largest single-center series describing long-term outcome included 819 patients (mean age at repair 17.2 ± 13.6 years) who underwent isolated operative repair of coarctation at Mayo Clinic between 1946 and 2005. The survival rates were 93%, 86%, and 74% at 10, 20, and 30 years after primary repair, respectively (58). In a previous report from this center, the most common cause of late death was coronary artery disease, followed by sudden death, HF, cerebrovascular accident, and ruptured aortic aneurysm (57).
Systemic Hypertension
Systemic hypertension is one of the major long-term problems following repair of coarctation of the aorta. Although the blood pressure typically falls after successful repair, persistent or recurrent hypertension and disproportionate systolic hypertension with exercise are observed, especially in patients whose repair is performed later in life. Although the blood pressure typically falls after successful repair, persistent or recurrent hypertension and disproportionate systolic hypertension with exercise are not uncommon. Hypertension and left ventricular hypertrophy are among the factors that contribute to premature death from coronary and cerebrovascular disease in patients with a surgically repaired coarctation (57).
The factors responsible for the persistent risk of hypertension after coarctation repair are not well understood. Among the probable contributing factors are structural and functional abnormalities that decrease compliance in the precoarctation arterial wall. Also, increased ventricular stiffness, left ventricular hypertrophy, and a hypercontractile state in postrepair patients may play a contributory role (59).
Multiple studies have found a significant incidence of systemic hypertension either at rest or with exercise following repair (60,61,62,63). When combining resting blood pressure, ambulatory blood pressure monitoring and exercise testing, systemic hypertension has been reported in as many as 70% of patients following coarctation repair (64). Hypertension may occur irrespective of the age at surgery or the presence of a residual gradient. Patients who had delayed initial repair often have residual hypertension despite surgical or transcatheter intervention. When hypertension is detected at rest, recoarctation must be excluded by physical examination (brachial/femoral pulse delay, arm/leg blood pressure gradient), Doppler echocardiography, and/or CMR or cardiac CT imaging. Recoarctation should be evaluated for transcatheter therapy (stent, angioplasty)—see adult congenital heart disease interventional therapy section.
If there is no evidence of recoarctation, then medical management for hypertension is indicated. It is important to control elevated blood pressure. As noted in the 2008 ACC/AHA adult congenital heart disease guidelines, hypertension should be controlled by beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin-receptor blockers (12). Once resting BP is normal, assessment of activity-related hypertension is performed with a 24 ambulatory BP monitor or in athletes, exercise study to determine peak systolic BP at maximal exercise.
Recoarctation
Recurrent recoarctation refers to restenosis after an initially successful intervention. Often the major findings suggest that a patient has developed recoarctation are resting hypertension and headaches, though some patients could still be asymptomatic. The rate of recoarctation is approximately 5% to 14% after surgery (65,66). It is seen primarily in children usually due to inadequate aortic wall growth at the site of repair when surgery is performed before the aorta has reached adult size. Following balloon angioplasty, children are also at greater risk for recoarctation compared with adults.
Recoarctation can be based on anatomic narrowing, arm/leg BP gradient, evidence of hypertension, or a combination of these factors (Fig. 67.9). Most patients with recoarctation will undergo an evaluation for transcatheter therapy to relieve the aortic obstruction (see section on adult congenital heart disease interventional therapy).
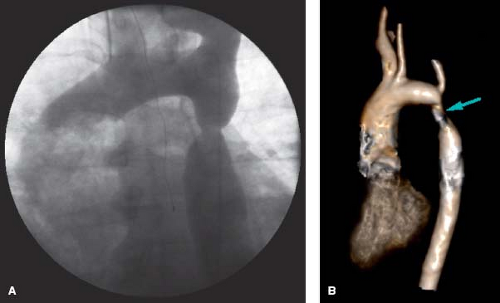 Figure 67.9 A: A 39-year-old patient with COA s/p end-to-end anastomosis presents with hypertension and is found to have recoarctation, subsequently relieved with transcatheter stent therapy. B: 3-D MRA showing the area of recoarctation. (B courtesy of Kan Hor, MD, Nationwide Children’s Hospital, Columbus, Ohio.) |
Indications for intervention for recoarctation include hypertension, a peak instantaneous pressure gradient across the coarctation ≥20 mm Hg, and/or imaging evidence of collateral circulation (67).
Discrete coarctation in older children and adults is treated with percutaneous balloon angioplasty, often with stent therapy (12,68,69). Eiken reported that despite successful stent therapy the patient may still demonstrate systemic hypertension requiring medical therapy, once again attesting to the intrinsic abnormality associated with coarctation of the aorta (70).
Aortic Aneurysm/Pseudoaneurysm
An aortic aneurysm may develop at the site of prior coarctation following surgery (especially after patch angioplasty), balloon dilation, or stent implantation of native coarctation (71). Development of aortic aneurysm and rupture may occur years after successful repair of coarctation of the aorta (39,40,41,42). Risk factors for postrepair aneurysms are age at the time of coarctation repair (≥13.5 years) and the use of patch angioplasty (72). The risk of dissection is increased during pregnancy, which is associated with hemodynamic, physiologic, and hormonal changes superimposed on the pre-existing aortic wall medial changes. This finding appears to occur without recurrent coarctation and despite relief of systemic hypertension (Fig. 67.10). For the majority of patients, aneurysm repair requires surgical intervention with resection of the aneurysm and graft placement. Alternatively, endovascular stent grafts have been used to repair aortic aneurysms at the site of prior coarctation repair. At this time, there are no criteria to guide the timing of aortic aneurysm repair in this population.
Pseudoaneurysms at the COA repair site can demonstrate an area of weakening with outpouching of the adventitial thin layer of the aorta, usually along the suture line. Pseudoaneurysms are at a higher risk for rupture and should be considered for repair at
the time of diagnosis. Either surgical repair or in selected cases, excluding the aneurysm with a covered stent should be employed to remove the risk of pseudoaneurysm rupture (Fig. 67.11).
the time of diagnosis. Either surgical repair or in selected cases, excluding the aneurysm with a covered stent should be employed to remove the risk of pseudoaneurysm rupture (Fig. 67.11).
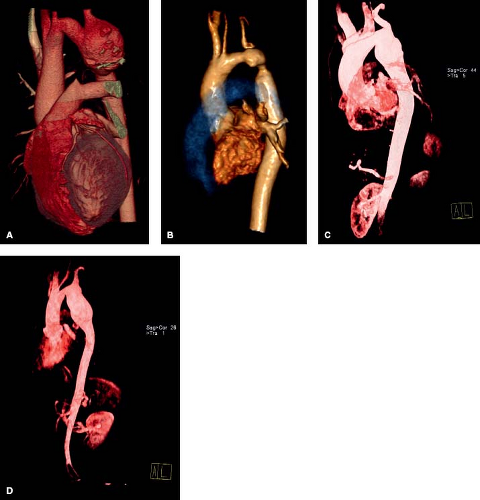 Figure 67.10 A: A 28-year-old asymptomatic patient s/p patch aortoplasty repair for COA presented for routine screening. Cardiac MRI demonstrated a large aortic aneurysm near the repair site. B: A 35-year-old s/p end-to-end anastomosis for COA presented with hypertension and back pain. CMR revealed recoarctation with a small aneurysm at the site of repair. C: A 38-year-old patient with end-to-end anastomosis COA repair, presents for routine screening found to have an aneurysm at repair site measuring 4.2 cm. D: A 48-year-old patient with patch aortoplasty repair for COA, upon routine screening found to have large 5.2-cm aneurysm. (B courtesy of Kan Hor, MD, Nationwide Children’s Hospital, Columbus, Ohio.) |
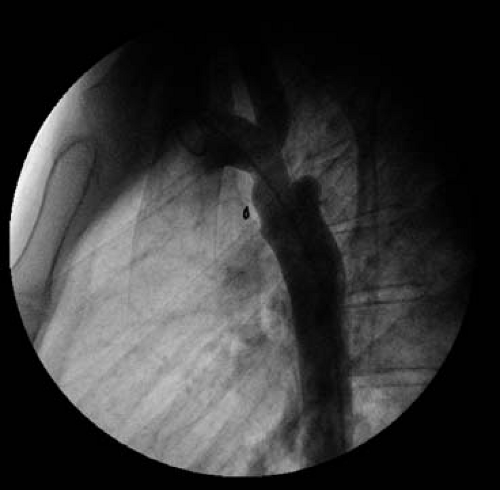 Figure 67.11 Aortogram from a 26-year-old patient with COA s/p end-to-end anastomosis found upon routine screening to have a small pseudoaneurysm and recoarctation at the repair site. The aneurysm was excluded and the obstruction relieved with a covered stent. |
Though risk factors for postrepair aneurysms have been identified, including a later age at initial repair and the use of patch angioplasty, there are no clear risk factors for the higher incidence of hypertension and aortic dissection following coarctation repair. However, a growing body of evidence demonstrates that there is an intrinsic abnormality of aortic function that persists despite adequate repair (43). Patients with repaired coarctation rarely can also develop aneurysms of the ascending aorta which often are associated with BAVs (73,74,75). A stiff or less distensible aorta has been described with essential hypertension, coronary artery disease and Marfan syndrome, and may be the underlying mechanism contributing to the late abnormalities associated with repaired coarctation of the aorta (76).
Documenting the type of repair performed is important in the evaluation of this population. Most underwent patch aortoplasty, resection of the coarctation with end-to-end anastomosis or subclavian flap repair. However, a small percent have undergone bypass tube grafting around the coarctation segment. A clear understanding of the type of repair will aid in the diagnosis of complications and when follow-up studies are necessary (Fig. 67.12). Therrien et al. demonstrated the most cost-effective evaluation for adults with repaired CoA includes a clinical visit (thorough history and physical examination) and a cardiac MRI to rule out aortic aneurysm and recoarctation (77).
Adult patients with previous coarctation repair should be followed serially for evidence hypertension, both at rest and with ambulatory monitoring, and should be carefully assessed for recoarctation, aortic aneurysms, and progressive valvular disease especially in those with concomitant aortic or mitral valve abnormalities.
Left-to-Right Shunting Lesions
Adults may present either with repaired or unrepaired hemodynamically significant shunt lesions including VSDs, atrioventricular septal defects (AVSD), ASDs, PDA, and in rare cases aorticopulmonary window, and arteriovenous (including coronary) fistulae. Those with shunt lesions repaired at a young age (VSD, AVSD, PDA <1 year) will be unlikely to develop pulmonary vascular disease. Large shunts either left unrepaired as a child or found incidentally as an adult will inevitably lead to elevated pulmonary vascular resistance (PVR) and Eisenmenger syndrome (ES).
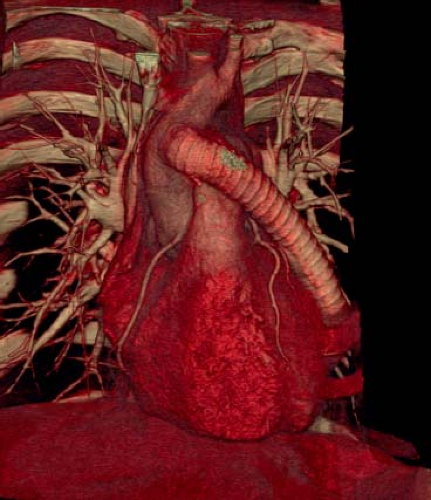 Figure 67.12 Cardiac CT in a 26-year-old patient with COA s/p ascending aorta to descending aorta tube graft. |
Ventricular Septal Defects
VSD is the most common congenital heart defect at birth, but accounts for only 10% of congenital heart defects in adults because many close spontaneously (78). Adult patients with VSD or AVSD associated with high pulmonary resistance (especially if operated later than 1 year of age) can develop progressive pulmonary vascular obstructive disease (PVOD) (79,80,81,82).
Patients with an isolated small restrictive VSD with small left-to-right shunt (often referred to as “maladie de Roger”) generally remain asymptomatic in adulthood, but AR may develop. Patients with moderate-sized VSDs may remain asymptomatic or develop symptoms of mild HF in childhood. Heart failure usually resolves with medical therapy and with time and the VSD becomes smaller in absolute and/or relative terms. Most patients with large VSDs have early large left-to-right shunting with the development of HF during infancy. In rare cases, the PVR does not fall normally postnatally, left-to-right shunting is less marked, and presentation with ES occurs sometime during late childhood to early adulthood. The right-to-left shunt causes cyanosis.
Many patients who have a VSD do not require repair because it is anatomically small, closing, or hemodynamically insignificant. This applies particularly to the perimembranous defect where septal aneurysmal tissue can partially occlude the defect and to some smaller muscular defects. On the other hand, false security might exist when a subpulmonic defect seems to be getting smaller. In these patients, an aortic cusp can prolapse and partially or completely
occlude the defect. In these patients, early operation is necessary to help protect the integrity of aortic valve coaptation.
occlude the defect. In these patients, early operation is necessary to help protect the integrity of aortic valve coaptation.
Though it is rare to find an adult patient who would benefit from isolated VSD closure, thorough clinical, echocardiographic assessment and at times invasive hemodynamic evaluations are important to determine the most appropriate timing of surgical or percutaneous VSD repair, to prevent the development of irreversible PVOD, and to preserve the integrity of the aortic valve and LV size and function. In unrepaired adults, either the VSD is small and restrictive and therefore closure would not be indicated, or the defect is large enough to have caused PVOD and closure would subsequently be considered harmful. Double-chambered right ventricle (DCRV) occurs only in association with membranous VSD, and if significantly obstructive, surgery is indicated.
The treatment of patients with VSD depends on the type of defect, its size, shunt severity, pulmonary artery (PA) pressure, vascular resistance, and associated acquired complications including DCRV, AR, and PHTN. Therefore, repair of VSD should be considered in all adult patients who are symptomatic or have signs of left ventricular volume overload without irreversible pulmonary vascular disease.
Indications for closure of a VSD in an adult are included in the 2008 ACC/AHA adult congenital heart disease guidelines as follows (67). Similar recommendations are included in the ESC and the Canadian Society of Cardiology guidelines (19).
Closure of a VSD is indicated when there is a pulmonary-to-systemic flow ratio (Qp/Qs) ≥2 and clinical evidence of left ventricular volume overload.
Closure of a VSD is indicated when the patient has a history of infective endocarditis.
Closure of a VSD is reasonable when there is net left-to-right shunting with a Qp/Qs ≥1.5 with PA pressure less than two-thirds systemic pressure and PVR is less than two-thirds systemic vascular resistance (SVR).
Closure of a VSD is reasonable when there is net left-to-right shunting with a Qp/Qs ≥1.5 in the presence of left ventricular systolic or diastolic dysfunction or failure.
Observation is reserved for asymptomatic patients with no evidence of left ventricular volume overload. Medical management is suggested for patients with symptoms and/or left ventricular volume overload who are not candidates for repair such as those with large defects and severe irreversible PVOD (Eisenmenger complex) (67). Survival in VSD patients ranges from excellent for those with small restrictive VSDs, normal ventricular function, and normal PA pressure to reduced for those with associated Eisenmenger complex.
Although primary surgical repair for isolated VSD was previously performed through a ventriculotomy, most are now performed through a right atriotomy as a first-line approach for most VSDs, transcatheter device VSD closure is a treatment option for isolated uncomplicated muscular VSDs, and for certain membranous VSDs, only in very selected patients who have suitable anatomy. The technical success rate of transcatheter closure of selected muscular and membranous VSDs is high and the mortality rate is low (83).
Hijazi et al. reported 10-year results of percutaneous closure in adult congenital and acquired (nonpost infarct) VSDs using different types of Amplatzer occluder devices. The patients ranged in age from 18 to 84 years with a median age of 34 years. Indications for closure included symptoms related to significant shunt (dyspnea on exertion), unexplained deterioration of LV function, and/or LV dilation, recurrent endocarditis, and concerns for PHTN. Not only did they show that the procedure was both safe (two minor procedural complications) but it was also very effective in eliminating intracardiac shunts and showing improvement in LV size after closure (84).
Long-term survival ranges from excellent for patients with small restrictive VSD, normal ventricular function, and normal PA pressures to limited for those with Eisenmenger complex.
Atrioventricular Septal Defects
AVSDs are anatomic defects that arise from faulty development of the embryonic endocardial cushions. This spectrum ranges from a primum ASD and cleft mitral valve, known as a partial AVSD, to defects of both the primum atrial septum and inlet ventricular septum and the presence of a common atrioventricular valve, referred to as complete AVSD. The terms atrioventricular canal defect and endocardial cushion defect are used in reference to this group of defects; however, AVSD is now the preferred terminology. Combinations of these anatomic abnormalities result in complete (both ASDs and VSDs) and partial (only ASD) forms that are manifested in varying clinical presentations.
Surgical correction for an AVSD is generally performed because of the significant morbidity and mortality associated with the uncorrected lesion. The management approach is based on the type of AVSD, determining whether there are other associated anatomic and hemodynamic abnormalities (e.g., relative ventricular size, AV valve regurgitation, and right or left ventricular outflow obstruction), and whether or not other cardiac conditions (e.g., PDA) are present (85).
Management of complete AVSDs encompasses initial medical management for HF, and either primary complete repair or palliative intervention. Patients with partial and transitional AV valve defects are generally asymptomatic during childhood, but are likely to develop symptoms of right heart overload as adults. Patients with significant left AV valve regurgitation are at risk for left HF and atrial fibrillation. As a result, we recommend that surgical correction be performed in patients with partial and transitional AVSD. The overall mortality rate for surgical repair of AVSD is about 3%, and the 10-year survival rate exceeds 90%. Morbidity associated with surgical repair includes postoperative PHTN and arrhythmias, and AV valve complications including regurgitation and, less commonly, stenosis. Patients with preoperative AV regurgitation are likely to have postoperative regurgitation, which often requires reoperation. Pulmonary overcirculation and congestive HF symptoms are common in the first several months of life as PVR continues to fall throughout infancy. In contrast, the adult with an uncorrected complete AVSD, the pulmonary vasculature is subjected to both increased blood flow and systemic pressure and eventually develops irreversible PVOD (ES).
A review of the Pediatric Heart Network data of primary surgical repair for AVSD performed prior to 1 year of age demonstrated both low mortality (3%) and a 20-year survival of about 95% (86,87). The most common residual lesion is left AV valve regurgitation. This finding is present in about 25% of patients as soon as 6 months after operation. Many of these patients may require reoperation. The current recommendations for reoperation include:
Left AV valve repair/replacement for regurgitation or stenosis causing symptoms, arrhythmias, LV dilation, or LV dysfunction.
LVOT obstruction (subaortic stenosis) with a mean gradient of more than 50 mm Hg or peak greater than 70 mm Hg. In the setting of mitral regurgitation (MR); surgical intervention should be considered with a gradient greater than 50 mm Hg.
Residual ASD or VSD that meets criteria for closure (12).
Patients with unrepaired defects in adulthood should undergo evaluation for PVOD. If PVR is acceptable, the patient should undergo complete repair of the defect. If PVOD is severe (ES) the patient should have close follow-up with specialists in adult congenital cardiology and PHTN. Advances in pulmonary vasodilator therapy have provided significant benefits with demonstrated improvements in functional capacity.
The ACC/AHA 2008 guidelines for the management of adults with congenital heart disease recommend that all patients with an AVSD should follow routinely with an adult congenital heart disease specialist. Most often patients should undergo annual evaluation (12). These guidelines can be tailored appropriately in the presence of residual sequelae such as AV valve regurgitation, subaortic obstruction, arrhythmias, and decreased ventricular function that develop after surgical repair or the presence of PHTN that warrants closer surveillance. Routine evaluation should include focused TTE that includes AV valve function, LVOT and ventricular function. Routine ECG and intermittent Holter monitor evaluation should be considered to evaluate for AV block (∼2%) as there is an increased risk of injury to the posteriorly displaced AV node during surgical repair.
In both the repaired and unrepaired patient, atrial arrhythmias can be common. In a study by Garson et al. of 380 pediatric patients, mean age 10.3 years, with atrial arrhythmias, 5% of these patients had an AVSD. Of these patients, 80% had a previous surgical repair (88). As atrial arrhythmias may contribute to a significant source of morbidity and mortality in this growing adult congenital population, the patient with AVSD (repaired or unrepaired) who presents with an atrial arrhythmia warrants a detailed evaluation of hemodynamics, specifically AV valve regurgitation, functional assessment, and residual defect (ASD/VSD). Associated concerns include residual shunts, aortic valve regurgitation, arrhythmias, including complete heart block requiring a pacemaker, PA deformity, or acquired pulmonary valve stenosis (PVS) from previously placed pulmonary arterial bands and residual atrioventricular valve regurgitation in the AVSD group. The varied constellation of symptoms can often make their clinical presentation, natural history, and treatment quite variable and treatment options are sometimes challenging.
Atrial Septal Defects
ASDs are the most common congenital lesion in adults after BAV. Although the defect is often asymptomatic until adulthood, potential complications of an undetected ASD include right ventricular failure, atrial arrhythmias, paradoxical embolization leading to a cerebrovascular accident or transient ischemic attacks, cerebral abscess, and PHTN that can become irreversible and lead to right-to-left shunting (ES).
Smaller ASDs close spontaneously in infants, however, spontaneous closure is unusual in older children and adults. Patients with ASDs are often operated before adulthood. Rarely patients will go undetected until a high school sports physical examination, or following an incidental chest film that is interpreted as abnormal. Exercise intolerance, fatigue, dyspnea, overt HF, and paradoxical embolization are manifestations of symptomatic ASDs in adults that warrant defect closure. Most patients do well in the long term after successful surgical closure and remain symptom free, though rare atrial arrhythmias or sick sinus syndrome can occur (89).
Echocardiography is the imaging modality of choice for the diagnosis of ASD. TTE is usually definitive for diagnosis of ostium secundum and primum defects (the latter generally accompanied by cleft mitral leaflet), while transesophageal echocardiography (TEE) may aid in the sizing of defects, the diagnosis of sinus venosus defects of superior vena cava (SVC) or inferior vena cava (IVC) type, and the assessment of associated congenital anomalies such as anomalous pulmonary venous connections. CMR imaging can determine ASD location and size, estimate shunt flow, and detect anomalous pulmonary venous connections. Cardiac CT may also be used to identify ASD location and size and detect anomalous pulmonary venous connections.
Once an ASD has been identified, the right heart should be assessed to identify any associated right atrial enlargement and right ventricular volume overload, since RV overload is the chief indication for ASD closure (12,13). Evidence of PHTN should be assessed in all patients with ASDs. Invasive cardiac catheterization is rarely required for diagnosis since the evaluation can generally be performed using noninvasive means. Cardiac catheterization is indicated if significant PHTN is present to determine the PVR and reversibility with vasodilator therapy.
Murphy et al. demonstrated that the age at operation and the PA pressure influences survival after ASD repair (90). Patients repaired <25 years of age have similar long-term survival compared to controls. However, patients ≥25 years old and those with PA systolic pressure ≥40 mm Hg at repair have a lower long-term survival apparently due to late cardiac failure, stroke, and atrial fibrillation (Fig. 67.13). The postoperative ASD patient should be followed,
albeit infrequently, with periodic examinations, electrocardiography and occasional 24-hour ambulatory monitoring.
albeit infrequently, with periodic examinations, electrocardiography and occasional 24-hour ambulatory monitoring.
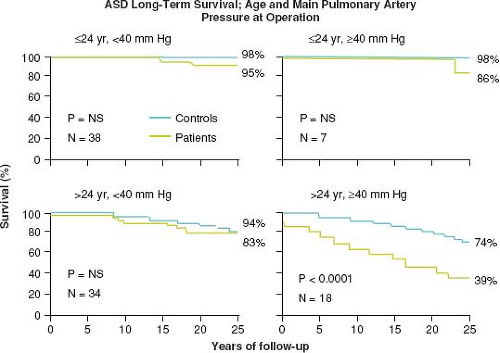 Figure 67.13 Survival curves for ASD patients based upon age at surgical repair (< or >24 years of age) and pulmonary artery systolic pressure at the time of repair (< or >40 mm Hg). Survival was significantly diminished only with a combination of age >24 years old and pulmonary artery systolic pressure >40 mm Hg (Kouchoukos NT, Dávila-Román VG, Spray TL, Murphy SF, Perrillo JB. Replacement of the aortic root with a pulmonary autograft in children and young adults with aortic-valve disease. N Engl J Med. 1994;330(1):1–6). |
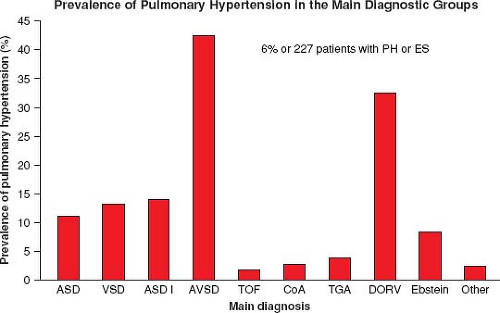 Figure 67.14 Prevalence of PH in the main diagnostic categories from the CONCOR project. (Data from Vander Velde, Vriend JW, Mannens MM, Uiterwaal CS, Brand R, Mulder BJ et al., CONCOR, an initiative towards a national registry and DNA-bank of patients with congenital heart disease in the Netherlands: rationale, design, and first results. Eur J Epidemiol. 2005;20:549–557.) |
Percutaneous device closure is an alternative to surgical closure in patients with ostium secundum ASDs that have appropriate anatomic characteristics. The United States Food and Drug Administration’s Center for Devices and Radiological Health has approved two devices for percutaneous ASD closure (57,58). These are the Amplatzer Septal Occluder and the CardioSEAL Septal Occlusion System. The indications for transcatheter device closure are the same as surgical closure (see Chapter 17) except that device closure with Amplatzer ASO is only indicated for secundum ASDs up to a maximum device size of 38 mm. Patients with sinus venosus or primum ASDs and any patient with ASD and anomalous pulmonary venous return require surgical repair.
Pulmonary Hypertension in Adults with Congenital Heart Disease
Pulmonary arterial hypertension (PAH), defined as a PA pressure of ≥25 mm Hg at rest in the presence of normal pulmonary capillary wedge pressure (i.e., ≤15 mm Hg), is relatively common among patients with congenital heart disease (91,92,93,94). PAH can develop in patients with left-to-right intracardiac shunts (ASDs and VSDs), especially when they are large and nonrestrictive, due to increased pulmonary blood volume or pressure overload. ES is the most severe and end-stage form of shunt-related PAH (see below). This group also includes patients that have PAH with coincidental or small defects and those with persistent or worsening PAH despite closure of the defect.
Significant left-to-right shunts unrepaired or those repaired at a later age may develop PHTN. Complex CHD lesions at risk to develop PH include:
Septal defects—ASD, VSD, AVSD, PDA
Single ventricle complexes—DORV, DILV
Transposition of the great arteries (TGA)
Truncus arteriosus
In a large ACHD registry from the Netherlands of over 5,000 patients, the overall incidence of PH was 4.2%, 6% in those with only a septal defect, and 10% of those with a risk high lesion for PH (categories above) (Fig. 67.14) (95,96). Of those with a septal defect and PH, 58% met the definition of ES. VSD was the most common diagnosis in the group with septal defects and PH; however, AVSD had the highest prevalence at 41% (Fig. 67.15).
A wide range of defects are often associated with PAH and patients with PAH–CHD represent a heterogeneous group. Gatzoulis et al. condensed these into the following four categories:
ES: Patients with left-to-right shunts due to large defects (atrial, ventricular or arterial, e.g., PDA, AP window), leading to severe PAH and, with time, to reversal of shunting. Chronic cyanosis with secondary erythrocytosis and multi-organ involvement are typically present in these patients. There is currently evidence to
support PAH-specific therapy for functional class III patients in this group.
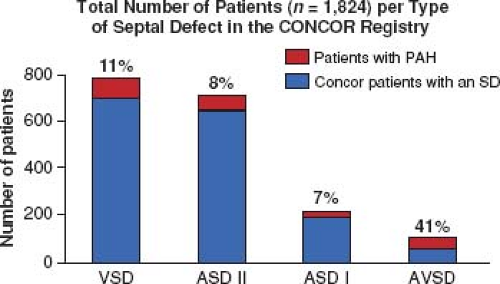
Figure 67.15 Total number of patients from the CONCOR project with septal defects and those with PH. VSD is the most common septal defect with PH and AVSD has the highest prevalence. (Data from Duffels MG, Engelfriet PM, Berger RM, et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int J Cardiol. 2007;120(2):198–204.)
PAH associated with left-to-right shunts: Patients with sizeable defects and mild-to-moderate PAH who shunt predominantly left to right and are not cyanosed at rest (i.e., patients who have not developed reversal of shunting). These patients have the potential to develop ES over time. Surgical or catheter interventions addressing the hemodynamic lesion in this setting are controversial and unlikely to convey long-term benefits if patients have elevated PVR (as hemodynamic intervention(s) do not seem to reverse established pulmonary vascular disease). The decision to intervene should not be based on the feasibility of the procedure. Treatment with PAH-specific therapy may have a role in this patient group, although controlled data are lacking at present.
PAH with small or coincidental defects: Patients with relatively small defects who have developed PAH that cannot be solely attributed to CHD. These patients should be treated with PAH-specific therapy and their small heart defects should not be closed.
PAH after surgical or percutaneous correction: Patients who present with progressive PAH early or late after repair of CHD in the absence of large residual hemodynamic lesions from the original operation. Such patients seem to have survival rates similar to patients with idiopathic PAH and worse than patients with PAH and open defects and even patients with ES (10). Consequently, these patients with PAH after reparative cardiac surgery for CHD should be treated with PAH-specific therapy (97).
Eisenmenger Syndrome
Patients with ES display chronic cyanosis and, typically, multi-organ, multi-system involvement. Chronic cyanosis has a detrimental effect on exercise capacity but it is also a powerful stimulus for secondary erythrocytosis which, in turn, increases oxygen-carrying capacity, enhancing tissue oxygenation and preventing, at least in part, hypoxic end-organ damage (98).
With the advent and subsequent advancement of surgical management of CHD, the incidence of ES is on the decline, though not negligible. The overall incidence of ES is not known. The diagnosis requires the presence of underlying CHD, though in some cases the diagnosis is not established until adulthood.
In the Netherlands study, 1% of all of the patients registered through their ACHD clinic and 58% of those with a septal defect and PH had ES (90). The development of ES is based upon the size and location of the left-to-right shunt. From the second natural history study of patients with VSD, 3% for those with a small-to-moderate defect (≤1.5 cm in diameter) will develop ES. However, with a large VSD (>1.5 cm), nearly 50% will develop ES (79). From the time PH diagnosis, ES survival is longer than those with the idiopathic PH and patients commonly survive into the third or fourth decade of life with ES. Patients with ES carry a survival rate of 80% at 10 years, 77% at 15 years, and 42% at 25 years. Poorer survival has been associated with syncope, elevated right heart filling pressures, and lower systemic saturations <85% (99,100).
ES patients are at risk for multiple medical problems including polycythemia, coagulopathy, especially platelet consumption, brain abscess, cerebral microemboli, hemoptysis, gout, and renal dysfunction (100). These patients should be routinely seen by centers with CHD–PAH trained physicians. The evaluation should include comprehensive assessment of their functional capacity, measurement of their hemoglobin, platelet count, iron studies, creatinine, and uric acid levels. Digital oximetry, both with and without supplemental oxygen therapy and oxygen-responsive hypoxemia should be investigated and they warrant expedited evaluation and treatment of underlying arrhythmias (101).
Other situations that are associated with a markedly increased risk in patients with ES include pregnancy, volume depletion, isometric exercise, high altitude, and endocardial pacing. Meticulous care of intravenous lines is required to avoid the risk of air or clot emboli. Pregnancy in patients with ES is associated with significant morbidity and mortality and is contraindicated. The reported rate of maternal mortality in patients with ES is reported at being between 30% and 50%, with pregnancy continuing to be prohibitive in such patients despite the advances in medical therapies (67). Gatzoulis et al. reported that the overall maternal mortality was lower compared with previous era (25% vs. 38%), with 17% mortality in patients with idiopathic PAH, 28% in those with CHD–PAH, and 33% in patients with other causes for PHTN (102).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


