Tetralogy of Fallot with Pulmonary Stenosis, Pulmonary Atresia, and Absent Pulmonary Valve
S. Lucy Roche
Steven C. Greenway
Andrew N. Redington
Tetralogy of Fallot (TOF) is a spectrum of diseases, sharing a common anatomic basis, but with varied presentation and management based on the exact anatomy of the right ventricular outflow tract. Consequently, some infants will be almost unaffected by their cardiac disease and undergo uncomplicated elective repair at a few months of age, while others are critically ill in the newborn period and represent a major therapeutic challenge. In many ways, TOF could be considered the archetype congenital heart disease (CHD); setting a pattern for the evolution of anatomical description, surgical management, pathophysiologic understanding, and the appreciation of late outcomes in adult life. The origins of congenital cardiac surgery can be traced back to the early palliative procedures for TOF, and the triumph of efforts toward its surgical “correction” were quickly replicated in other conditions. Over the past 60 years, outcomes for patients with TOF have been transformed, so that now (assuming they have access to surgery) almost all infants born with TOF are expected to survive. Through the careful long-term study of these patients, TOF has become a model for late pathophysiology after CHD repair, and as we have learned more, our approach to surgical correction has adapted and changed. The late outcomes of patients with repaired TOF taught us the importance of providing lifelong follow-up for patients with CHD, and at the other end of the spectrum, TOF is leading the way in elucidating the genetic causes of CHD. In this chapter, we review not just the epidemiology, anatomy, and treatment of TOF, but its history. We hope to highlight just how much progress has been made and how the contributions of many have revolutionized outcomes for patients with this condition and developed our understanding of CHD as a whole.
Epidemiology
TOF is the commonest form of cyanotic CHD. A 2002 meta-analysis of the incidence of CHD, which included 41 studies pertaining to TOF, suggested that the best estimate of incidence would be 577 cases of TOF per million live births (1). The prevalence of TOF (number of patients living with the diagnosis at any given time) has significantly increased since the introduction of successful surgical repair. In the year 2000, the prevalence of TOF in the adult population of Quebec was ≈0.17/1,000, and in Quebec’s children, the prevalence was ≈0.49/1,000 (2). Consequently, as outcomes continue to improve, it has been suggested that for many countries there are now numerically more adults with TOF than children.
TOF has a nearly equal sex distribution with perhaps a slight predominance in males. In a study that included information from 2.5 million live born infants and stillborn fetuses over a 10-year period, the Californian Birth Defects Monitoring Program reported a 1.1 relative risk of TOF in males compared to females (CI 0.9 to 1.3) (3). Maternal race seems to have little effect on incidence (4). Several studies have considered recurrence risk, both in siblings and offspring of the proband. A patient with TOF has approximately 2% to 3% chance of having a sibling with CHD, although not necessarily TOF (5,6). Recurrence risk in the offspring of patients with TOF is undoubtedly affected by whether or not the proband has 22q11.2 deletion. In the absence of 22q11.2 deletion, prospective parents with TOF might expect a 3% to 4% risk of having a child with CHD (5,7).
Etiology (Genetic and Environmental Factors)
Although familial disease contributes only a small fraction to the overall incidence of TOF, its increased frequency in consanguineous populations (8) and markedly increased recurrence risk in some pedigrees (9) allude to a central role for genetics in the underlying etiology. While a chromosomal duplication or a microdeletion at the 22q11.2 locus occurs in approximately 20% of patients with TOF and single gene mutations have been identified in others, in at least 50% to 60% of patients with TOF the causal genetic influences remain unknown (Fig. 41.1). However, this is a rapidly advancing field and with advances in both genomics and our understanding of human cardiogenesis, this percentage is likely to decrease. The relatively large number of distinct genetic loci that have been associated with TOF highlights the complexity and apparent genetic heterogeneity of cardiovascular development.
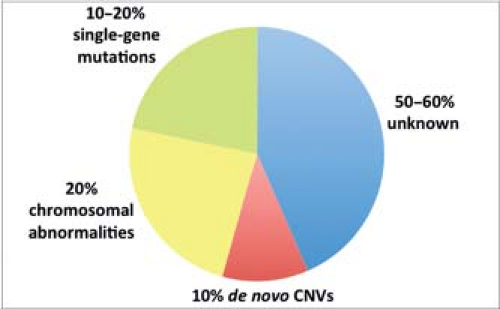 Figure 41.1 An illustration of the genetic causes of TOF. |
TOF is found in two broad categories of patients: syndromic TOF, which occurs in the presence of additional noncardiac congenital anomalies and, the more common, nonsyndromic TOF, which occurs in apparent isolation, usually appearing sporadically in families with no history of CHD. Even within these two distinct
classes, there is significant genetic overlap with mutations having been identified in the same gene in both syndromic and nonsyndromic patients (Fig. 41.2).
classes, there is significant genetic overlap with mutations having been identified in the same gene in both syndromic and nonsyndromic patients (Fig. 41.2).
 Figure 41.2 Genetic mutations identified in patients with syndromic and nonsyndromic TOF. |
Syndromic Tetralogy of Fallot
To date, TOF is associated with 121 entries in the Online Mendelian Inheritance in Man (OMIM) database (http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/omim), and there are 32 listed syndromes that include the presence of TOF as a characteristic feature. Furthermore, there are many case reports and series of TOF in association with other syndromes in which the presence of a heart defect is more variable. The best known syndrome and most frequently identified cause of TOF occur as a result of a 22q11.2 microdeletion. This deletion has a prevalence of approximately 1 per 6,000 to 10,000 live births, with males and females equally affected (10,11,12), although frequencies as high as 1 per 3,000 to 1 in 6,000 live births have been cited in some populations (13). A hemizygous deletion of chromosome 22q11.2 or, rarely, point mutations within TBX1 itself cause haploinsufficiency of the transcription factor TBX1 and result in the 22q11.2 microdeletion syndrome also known as DiGeorge syndrome, the velocardiofacial syndrome, or by several other names (14,15,16,17,18). Today, the recommendation is that the syndrome be described using molecular terminology, that is, 22q11.2 microdeletion with the term DiGeorge syndrome reserved for those rare patients who have the clinical features but with no identified mutation (13).
The most common mutation at the 22q11.2 locus is a 3 megabase (Mb) deletion responsible for 90% of cases (19). Atypical deletions have also been described and increasing use of SNP arrays as an alternative to fluorescent in situ hybridization (FiSH) for molecular diagnosis may enhance our knowledge regarding the frequency and effect of these atypical mutations (13). While most cases of 22q11.2 deletion arise sporadically as the result of de novo mutations, the inheritance of defects at this locus is autosomal dominant. Currently, 6% to 10% of new cases are familial, a percentage that is likely to increase due to the survival of individuals with previously lethal CHD (13). Since the phenotype is highly variable, even within families, it is not uncommon that genetic screening of a proband’s family identifies the microdeletion in parents or siblings with mild or no clinical evidence of the disorder.
22q11.2 microdeletion syndrome is characterized by the presence of a conotruncal defect (TOF, pulmonary atresia with ventricular septal defect [VSD], persistent arterial trunk, interrupted aortic arch, isolated arch anomalies, and VSD), immunodeficiency, neonatal hypocalcemia, developmental or psychiatric abnormalities, facial dysmorphisms, and palatal defects (13). However, its precise features are highly variable. In a cohort of 251 patients with conotruncal lesions, a chromosome 22q11.2 deletion was found in 16% of TOF patients, 50% of patients with interrupted aortic arch type B, 35% of patients with persistent arterial trunk, and 33% of patients with an outlet (malalignment) VSD (20). The likelihood of finding a 22q11.2 deletion in the presence of a conotruncal defect is further increased by the presence of anomalies of the aortic arch (right-sided or high arch), subclavian artery, pulmonary artery (PA) (atresia, absent central confluence, absent pulmonary valve, or multiple aortopulmonary collateral arteries), or absence of the arterial duct (14,21). Perhaps unsurprisingly, the clinical outcomes for patients with TOF and pulmonary atresia are worse when associated with 22q11.2 deletion (22). Interestingly, 22q11.2 deletion is very rarely found in association with other forms of CHD, such as transposition of the great arteries and hypoplastic left heart syndrome (10).
The next most common causes of syndromic TOF are those associated with major chromosomal abnormalities including Down syndrome (Trisomy 21), Edward syndrome (Trisomy 18), and Patau syndrome (Trisomy 13). These trisomies are estimated to cause 5% to 7% of syndromic TOF with Trisomy 21 responsible for most cases (23).
Smaller deletions and duplications have also been described as causes of syndromic TOF. These copy number variants (CNVs) are larger than 1,000 base pairs but are submicroscopic and, therefore, not detected on a routine karyotype. The importance of these rare variants in human disease has recently been appreciated due to technologic advances allowing genome-wide surveys at greater resolution than previously possible. These CNVs identify known or candidate TOF disease genes (24,25). At the current time, these CNVs account for only a small fraction of syndromic TOF but, with increasing use of microarrays and high-throughput DNA sequencing, we can expect to identify genetic abnormalities in a larger proportion of TOF patients. Microarray testing for CNVs in the population of syndromic TOF patients may have a relatively high yield since approximately 25% of patients with syndromic CHD have an identifiable mutation (25,26).
Single gene causes of syndromic TOF have been described and include mutations in TBX5 causing Holt–Oram syndrome, which manifests most commonly as forearm and thumb anomalies with atrial septal defect (27). Mutations in JAG1 and NOTCH2 cause Alagille syndrome, a disorder characterized by cholestasis, CHD, skeletal and ocular anomalies, and characteristic dysmorphic features (28,29,30).
Nonsyndromic Tetralogy of Fallot
Mutations in many genes have been linked with nonsyndromic TOF (see Fig. 41.2). In some cases, gene discovery in syndromic patients has been helpful in identifying the cause of nonsyndromic TOF. For example, 22q11.2 deletion may be present in up to 6% of patients with nonsyndromic TOF and appears more frequently in those with pulmonary atresia (31). In addition, mutations in JAG1, initially identified in Alagille syndrome, have been found in patients with isolated TOF (32,33,34,35), and mutations causing nonsyndromic TOF have also been identified in TBX1, which is deleted in the 22q11.2 microdeletion syndrome (23,32). Few studies of nonsyndromic TOF have considered inheritance; in those that have, mutations were inherited from phenotypically normal parents. This observation suggests reduced penetrance and/or the presence of additional cofactors and has led to the proposal that nonsyndromic TOF may fit a “multiple-hit” model of etiology (5,36). A variety of environmental exposures have been identified that contribute a modest increased risk for TOF or other conotruncal defects and include maternal pregestational diabetes, febrile or viral illnesses, vitamin A exposure, and exposure to organic solvents (37). Perhaps the development of nonsyndromic TOF requires both genetic susceptibility and one or more
environmental insults. However, to date, evidence supporting this hypothesis remains weak and anecdotal.
environmental insults. However, to date, evidence supporting this hypothesis remains weak and anecdotal.
The contribution of CNVs to nonsyndromic TOF has also been examined (34,38). Similar to the loci identified in syndromic TOF, these CNVs identify known and putative TOF disease genes. Of the 12 CNVs identified, there were only three loci affecting known disease genes (JAG1, NOTCH1, and TBX1). Furthermore, several of these loci affected regions of DNA that did not contain any known genes, suggesting that mutations in non–protein-coding DNA may contribute to the development of disease. The most common CNV affected the 1q21.1 locus, which accounted for 1% of TOF cases in this study (34). This locus has been identified by other groups and has been associated with psychiatric and neurologic disease as well as TOF and other CHD (39,40,41,42,43,44,45,46,47,48). This potential link between “nonstructural” nervous system disease and TOF is certainly intriguing since it parallels the findings for the 22q11.2 deletion and syndromic TOF.
To date, mutations in 12 single genes have been associated with nonsyndromic TOF but, thus far, each mutation appears to contribute to only a tiny percentage of cases. The disease genes identified primarily consist of transcription factors (NKX2.5 (49,50), GATA4 (51,52,53), ZFPM2 (54,55), GATA6 (56,57), TBX1 (23,58), and TBX20 (59)) and receptors or ligands in the NOTCH (NOTCH1 (60) and JAG1 (23,33)) or NODAL (FOXH1 (61), TDGF1 (61), GDF1 (62), and CFC1 (61)) pathways.
Current and Future Roles of Genetic Testing and Counseling in Tetralogy of Fallot
The potential genetic links and implications of genetic testing in TOF should be discussed with the parents of each newly diagnosed patient, whether a diagnosis of TOF has been made in a fetus or postnatally. There is also a role for genetic counseling in older patients, who may have been diagnosed with TOF at a time before much was known about its potential genetic etiology. In each case, the patient or parents should decide whether or not genetic testing is appropriate after considering the potential benefits and disadvantages. When combined with adequate resources for counseling, genetic testing can provide useful information for physicians, the parents of children with TOF, and the patients themselves.
For physicians, genetic testing of patients with TOF may identify a syndrome associated with other abnormalities that should be considered when planning management and discussing prognosis. For instance, there are potentially important reasons to identify a 22q11.2 deletion in infants with TOF before they undergo cardiac surgery (63). First, at the time of surgery and during the postoperative period, it is useful to anticipate and correct hypocalcemia, which can result as a consequence of the hypoparathyroidism seen in over 50% of patients with 22q11.2 deletion (64). Second, a small percentage of infants with 22q11.2 deletion exhibit complete T-cell deficiency, a condition known as “complete DiGeorge syndrome.” Such patients are at risk of iatrogenic graft-versus-host disease (GVHD), which may occur due to transfusion of lymphocyte-containing blood components and is a potentially fatal, but preventable, complication (65). Transfusion-associated GVHD is usually only a risk if CD4 and CD8 T-cell counts are below 1,000 and 800 cells/mm3, respectively (66). Guidelines suggest that infants with known or suspected 22q11.2 deletion should receive irradiated blood products at the time of cardiac surgery unless T-cell lymphocyte deficiency has been specifically excluded (67).
For the parents of a child with TOF, and for the patients themselves, another possible benefit of genetic counseling and testing is the provision of information that might inform future reproductive decisions. In addition, genetic testing may identify a condition that is associated with learning difficulties. In such cases, obtaining a genetic diagnosis may help the family of a patient with TOF obtain additional educational support from an early age. As they age, patients with 22q.11 microdeletion remain at risk of symptomatic hypocalcemia and may also develop psychiatric disorders that would benefit from early recognition and treatment (68).
Recommendations for genetic testing in TOF and other forms of CHD have been developed (21). Currently, these guidelines advocate genetic testing only in children with syndromic TOF (21). However, many centers have already adopted routine genetic screening for all patients with TOF. Although routine clinical genetic testing is currently limited to the detection of large chromosomal abnormalities either through karyotyping or through FiSH, advances in technology and decreasing costs in DNA sequencing may lead to an increased prevalence of this approach together with improved diagnostic yields. Furthermore, approximately 6% to 10% of children with a 22q11.2 deletion will have no other features of the disorder and will be missed if screening is confined to those only with obvious external abnormalities.
Anatomy (Morphologic, Echocardiographic, and Angiographic Correlations)
The journey toward anatomic understanding of this complex heart defect began well before the time of Etienne-Louis Arthur Fallot and continues to this day. In his 1,888 papers concerning “la maladie bleue,” Fallot acknowledged earlier reports of patients with the same pathology, including the paper by Danish anatomist and Bishop, Niels Stenson, which is considered to be the earliest description (69). In 1671, Stenson reported his pathologic findings in a fetus with multiple abnormalities, including cardiac features we would now recognize as TOF (70). One hundred years later, Eduard Sandifort, an anatomist at the University of Leiden, published the first clinical–anatomical correlate of this condition when he described the life and subsequent autopsy findings of a 12-year-old boy who had suffered from progressive cyanosis and breathlessness despite being “perfectly normal at birth” (71). Detailed anatomical diagrams accompanied his report and, unlike others present at the autopsy, Sandifort realized that the condition must have been congenital (71). In the 1800s, there were other reports including 15 cases published in John Farre’s textbook on cardiac malformations (72) and 64 cases referenced by Thomas Bevill Peacock (73). Fallot’s important contribution was to clearly define the clustering of four distinct anatomical features as a frequent cause of cardiac cyanosis and to introduce the term “tetralogy.” This defining contribution was forever acknowledged when Dr. Maude Abbott gave the disease its eponym. Fallot also dispelled the widely held belief that the blue discoloration of these patients was due to patency of the oval fossa (74). Fallot’s tetralogy, as described by him, is: stenosis of the PA, a VSD, hypertrophy of the right ventricle (RV), and rightward deviation of the origin of the aorta (69); and, for over 120 years, generations of medical students have memorized these cardinal features.
More recently, increasing knowledge of embryonic growth, understanding of cardiac morphology, and the development of rigorous nomenclature systems have resulted in a search for the pathognomonic features unique to a diagnosis of TOF. In today’s perception, TOF is generally considered a “monology” from which all four characteristic features result. The two great schools of cardiac morphology have somewhat different perspectives on this matter but the areas of controversy are relatively minor. In Anderson’s view, the characteristic abnormalities of TOF occur because the components that normally unite to form the outlet of the RV remain separate during development. He maintains that the defining morphology is anterior-cephalad deviation of the outlet (infundibular) septum (which may sometimes be only a fibrous remnant) relative to the septomarginal trabeculation (SMT) together with malformation of the SMTs (75,76). In contrast, the Van Praaghs judge underdevelopment of the
subpulmonary infundibulum to be the primary pathology; with all characteristics of TOF (including anterior-cephalad deviation of the outlet septum) being sequelae of this one pathologic feature (77,78). Whichever abnormality ultimately proves to be defining, anterior-cephalad deviation of the outlet septum (which is a much larger structure in TOF than in the normal heart) is certainly a cardinal feature. Since deviation of the outlet septum is often easily appreciated by echocardiography, identification of this anatomy is key to diagnosis (Fig. 41.3). No matter which the school of thought, the various forms of the disease discussed in this chapter can all be correctly discussed under the diagnostic umbrella of TOF, with additional characteristics (e.g., RVOT patency or atresia, absent pulmonary valve syndrome, with or without double outlet connection) all associated features, albeit profoundly important ones that define the presentation and management.
subpulmonary infundibulum to be the primary pathology; with all characteristics of TOF (including anterior-cephalad deviation of the outlet septum) being sequelae of this one pathologic feature (77,78). Whichever abnormality ultimately proves to be defining, anterior-cephalad deviation of the outlet septum (which is a much larger structure in TOF than in the normal heart) is certainly a cardinal feature. Since deviation of the outlet septum is often easily appreciated by echocardiography, identification of this anatomy is key to diagnosis (Fig. 41.3). No matter which the school of thought, the various forms of the disease discussed in this chapter can all be correctly discussed under the diagnostic umbrella of TOF, with additional characteristics (e.g., RVOT patency or atresia, absent pulmonary valve syndrome, with or without double outlet connection) all associated features, albeit profoundly important ones that define the presentation and management.
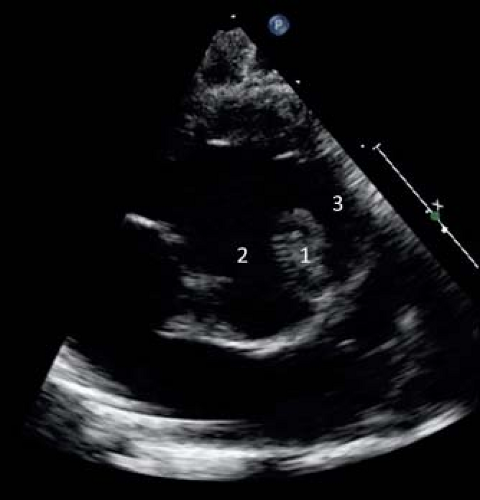 Figure 41.3 Parasternal short axis view of an infant with TOF demonstrating: (1) anterior and cephalad deviation of the outlet septum, (2) the VSD, and (3) narrowing of the subpulmonary RV outflow tract. |
The Interventricular Communication
The large, unrestrictive interventricular communication of TOF is always “roofed” by the aortic valve (Fig. 41.4) but its remaining borders are variable and sometimes difficult to define since they depend on the tissue planes considered. In general, the defect is described from its RV aspect (which is the aspect the surgeon encounters when closing it). The crest of the ventricular septum usually terminates with the Y-shaped division of the anterior and posterior limbs of the SMT. In approximately one-fifth of Caucasian patients, a muscular rim, formed by the limbs of the SMT, the ventriculoinfundibular fold (VIF), and the muscular outlet septum, encircles the RV side of the VSD posteroinferiorly and thus protects the conduction axis from damage (75). More commonly, the VIF fails to reach the SMT and an area of fibrous continuity exists between the aortic cusps and the septal leaflet of the tricuspid valve to form the posteroinferior margin of the defect. In these cases, the defect incorporates a remnant of the membranous septum and is perimembranous (75). Its superior margin is usually formed by the deviated muscular outlet (or infundibular) septum which, when associated with pulmonary atresia, may fuse with the anterior limb of the SMT. In other forms the atresia is not muscular, the narrow outflow tract terminating in an atretic pulmonary valve (see below).
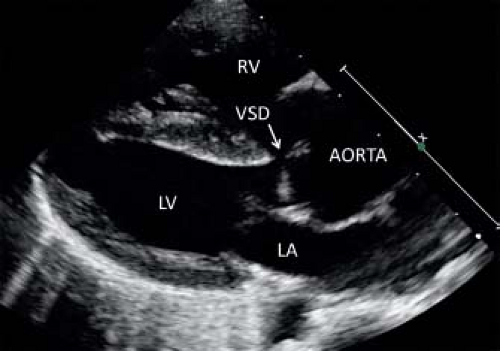 Figure 41.4 Typical parasternal long-axis view in a patient with TOF. The aorta overrides the crest of the ventricular septum. VSD, ventricular septal defect; LV, left ventricle; RV, right ventricle; LA, left atrium. |
 Figure 41.5 Subcostal right oblique section of TOF with a doubly committed VSD. The arrow points to point of fibrous continuity between the leaflets of the aortic valve and the hypoplastic pulmonary valve. RV, right ventricle; Ao, aorta; PT, pulmonary trunk. |
In some patients, there is absence, or near absence, of the infundibular septum, and the cusps of the aortic and pulmonary valves are in fibrous continuity and together form the superior border of the VSD. In these cases, the VSD is described as both doubly committed and subarterial, and while usually there is a muscular posteroinferior rim, the defect may also be perimembranous. This variant is rare in some populations, but relatively frequent in those of South American or South-East Asian descent (Fig. 41.5). In the past, it was argued that such patients should not be classified as having TOF since they have no muscular infundibular septum. It has been argued, however, that the outlet septum is present, albeit in the form of a fibrous remnant or raphe (79) and, since postnatally these patients exhibit all four classical features, this entity is now accepted to be a variant of TOF (76,80). TOF with a doubly committed VSD may be diagnosed preoperatively by echocardiography
or angiography (79,81). Compared to the majority of patients with TOF, those with a doubly committed VSD are at higher risk for the development of aortic regurgitation, both before and after repair, and have a higher incidence of residual postoperative RVOT obstruction (RVOTO) (82).
or angiography (79,81). Compared to the majority of patients with TOF, those with a doubly committed VSD are at higher risk for the development of aortic regurgitation, both before and after repair, and have a higher incidence of residual postoperative RVOT obstruction (RVOTO) (82).
The large outlet VSD seen in TOF can extend into the inlet component of the ventricle, either with or without an associated common atrioventricular valve, and there may be additional muscular VSDs that can be difficult to image by echocardiography because of the equalization of RV and left ventricle (LV) pressures. In each case, effort should be made to exclude additional muscular defects by imaging the ventricular septum with reduced Doppler color scale.
Aortic Position
In TOF, if an imaginary line were drawn perpendicular to the long axis of the heart, upward from the crest of the ventricular septum, it would transect the cusps of the closed aortic valve and, when the valve opened, this line would extend onward into the aortic root. This is what is meant by the term “overriding aorta.” In patients with TOF, the aorta overrides the VSD, a relationship best appreciated by echocardiography from the parasternal long-axis view (see Fig. 41.4). The degree of aortic override varies significantly between 15% and 95% (83), a feature which has created some confusion. Contention remains as to whether or not hearts in which there is >50% aortic override should be classified as double outlet RV, irrespective as to whether there is a bilateral infundibulum. The precise mechanics underlying aortic override in TOF remain incompletely understood. However, most authorities agree that, compared to normal, there is rightward malposition and clockwise rotation of the aortic root (77,78,84,85,86).
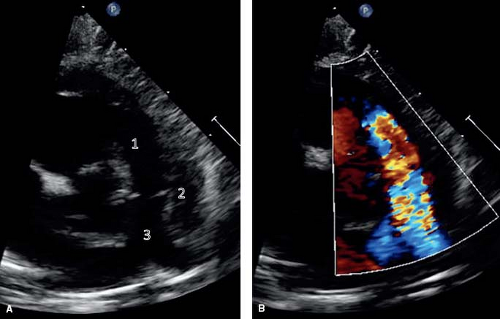 Figure 41.6 Parasternal short axis sections showing anatomic features (A) and corresponding color-flow Doppler signal (B). There is potential for multilevel obstruction of the RV outflow tract. This can occur because of (1) subpulmonary infundibular obstruction, (2) valvular pulmonary stenosis, and (3) branch PA stenosis. The RVOT is best imaged in a parasternal short-axis view. Color flow Doppler can be used to demonstrate both the location and severity of obstruction. |
Right Ventricular Outflow Tract Obstruction
Multilevel obstruction to pulmonary blood flow is a hallmark of TOF although the severity of obstruction varies considerably from patient to patient (Fig. 41.6). The typical pulmonary valve in TOF is thickened and obstructive, and is frequently bicuspid. The length of the infundibular region is similar to that of normal hearts (77,84), but its diameter is usually significantly reduced. Whether this reduction in diameter is primarily due to developmental hypoplasia (77,78) or is caused by anterior-cephalad deviation of the outlet septum combined with hypertrophy of the SMTs (75) remains of academic interest but, from the patients’ perspective, it is the effect of this narrowing that is important. The degree of narrowing of the subpulmonary infundibulum is variable in extent and timing. Progression from a patent RVOT to atresia during fetal life is well described, resulting in TOF with PA (87). While the latter is frequently referred to as pulmonary atresia with VSD, that does not do justice to the common fundamental anatomy that describes the spectrum or “family” of tetralogy lesions, and is also imprecise. There are many lesions that are characterized by a combination of pulmonary atresia and a VSD (e.g., functionally univentricular hearts, congenitally corrected transposition), but their anatomy and management are quite different. Importantly, the distal pulmonary arteries are frequently abnormal in TOF. Supravalvar stenosis is common, and the main PA and its branches exhibit highly variable
anatomy with important abnormalities in up to 40% of patients with a patent RVOT (88,89). The high incidence of discrete stenosis at the origin of the left PA is related to constriction of a sling of ductal tissue at the site of its insertion (88).
anatomy with important abnormalities in up to 40% of patients with a patent RVOT (88,89). The high incidence of discrete stenosis at the origin of the left PA is related to constriction of a sling of ductal tissue at the site of its insertion (88).
Abnormalities of the branch PAs are much more frequent in those with an atretic outflow tract, and while major aortopulmonary collateral arteries (MAPCAs) are not exclusive to those with pulmonary atresia, they are much more frequent, and are almost always associated with arborization abnormalities of the native PAs. The extent of PA atresia is quite variable and can involve the central pulmonary arteries (i.e., those outside the lungs) either proximally and/or distally. The atretic arterial segment can be recognized as a solid elastic cord in about 75% of the cases but is unidentifiable in the other cases. Most commonly, the pulmonary valve and the proximal portion of the pulmonary trunk (PT) are involved. It is important to recognize nonconfluence of the pulmonary arteries, and the ductal supply associated with such an arrangement, as it is rare to see MAPCAs and ductal supply to the same lung (see below).
Aortopulmonary Collaterals
Collateral vessels from the aorta to the pulmonary arteries are unusual in TOF in the absence of pulmonary atresia but approximately 6% of patients have a patent arterial duct (88,89).
The central right and left pulmonary arteries and/or the segmental pulmonary arteries can be confluent or nonconfluent. In tetralogy with pulmonary atresia, the blood supply to the lungs is entirely from the systemic arterial circulation, although occasionally the anatomy of the pulmonary arteries can be nearly identical in the presence of a pinhole orifice in the RVOT. The sources are the ductus arteriosus, systemic-to-pulmonary collateral arteries (henceforth designated simply as collateral arteries), occasionally a coronary artery, and plexuses of bronchial or pleural arteries (90). Ductal and collateral sources may coexist in the same patient but only rarely coexist in the same lung (90). A single systemic arterial source to a lung is termed a unifocal blood supply, whereas multiple sources are termed multifocal blood supply.
The caliber of the central pulmonary arteries varies considerably and appears to be directly related to the amount of flow (90). When the ductus or collateral arteries connect proximally to the central pulmonary arteries or their lobar branches, the central vessels may be only mildly hypoplastic or even normal in size. In contrast, when multiple collateral arteries anastomose more distally at segmental or subsegmental levels, the central pulmonary arteries tend to be quite hypoplastic. Furthermore, stenosis of the systemic arterial channels, either congenital or acquired, can be associated with hypoplasia of the central pulmonary arteries (90).
The patterns of intrapulmonary arterial distribution may be quite complex and are determined primarily by the types of systemic arterial supply (90). When a ductus supplies confluent central pulmonary arteries, the intrapulmonary arteries of both lungs are normal. When nonconfluent central pulmonary arteries are present, however, the lung supplied by a ductus will have a unifocal blood supply and a normal arterial distribution, but the contralateral lung usually has a multifocal blood supply and variable central pulmonary arterial distribution (90). When both lungs are supplied by multiple collateral channels, intrapulmonary arborization abnormalities are the rule (90), and a ductus is absent.
The ductus arteriosus, when present, usually is a unilateral structure and is associated with confluent pulmonary arteries in >80% of cases (90). Rarely, bilateral ducts may occur with nonconfluent pulmonary arteries. Unlike collateral arteries, the ductus does not branch before joining the central pulmonary arteries, and it tends to be less tortuous than collaterals. Because the ductus is widely patent during fetal life, the pulmonary arteries may be of normal size at birth. However, normal postnatal ductal narrowing usually occurs and produces distal stenosis in 35% to 50% of cases (90). As a result, blood flow to the lungs is diminished, and relative hypoplasia of the pulmonary arteries becomes more severe as the child grows.
Collateral arteries, when present, arise most commonly from the descending thoracic aorta, less commonly from the subclavian arteries, and rarely from the abdominal aorta or its branches or from the coronary arteries (90,91,92). Their number varies from 1 to 6, and their diameter ranges from 1 to 20 mm (90). Stenoses are present in nearly 60% of collateral arteries and tend to occur near the aortic or intrapulmonary anastomoses. The stenoses may be discrete or segmental, congenital or acquired (90). Anastomoses between the central pulmonary arteries (or their branches) and the collateral arteries are observed in about 40% of patients and may occur at the hilum or within the lung (90,93). In the remaining 60%, the collateral arteries enter the pulmonary hilum, travel with the bronchi as pulmonary arteries, and supply a variable number of bronchopulmonary segments (90). Although the central pulmonary arteries are confluent in about two-thirds of the cases, they usually supply only a portion of each lung owing to the coexistence of multiple collateral arteries and arborization abnormalities (90).
Small arterial plexuses that follow the bronchi or spread over the pleural surfaces may be apparent angiographically in more than half of the patients (90). They may arise from the aorta and its thoracic branches or from the systemic collateral arteries (90). It is thought that these vessels enlarge and proliferate postnatally in response to regional reductions in blood flow.
The ductus arteriosus is a precarious sole source of pulmonary blood supply, and its tendency to close necessitates surgical intervention in early infancy in more than half of the cases. Collateral arteries appear to be a more stable source of pulmonary flow, presumably because of multiple numbers. However, stenosis of collaterals may develop progressively, and they may become inadequate as the patient grows. Furthermore, because of hyperperfusion of some bronchopulmonary segments and hypoperfusion of others in the same patient, the pulmonary vascular bed may exhibit various histopathologic lesions, ranging from hypertensive pulmonary vascular disease to stasis thrombosis (90).
Coronary Arteries
The prevalence of coronary artery abnormalities in TOF varies somewhat between surgical, pathologic, and angiographic series but is approximately 5% to 7% (94). The significance lies mainly in an increased vulnerability of the anomalous vessels to injury during surgical repair, particularly if they cross the RVOT (95,96,97). In approximately 4% of TOF patients, the left anterior descending artery (LAD), or an accessory LAD, takes origin from the right coronary artery (RCA) or right coronary sinus and crosses the RVOT in its course toward the LV (94). A single coronary artery (more often from the left coronary sinus) is the second most frequent variant and occurs in ≈1% of patients (94). The solitary artery usually divides early into left and right branches, one of which may pass behind or in front of the RVOT. When a single coronary arises from the left sinus, it is the right branch that lies in a vulnerable position, whereas when the single coronary arises from the right sinus, it is the left branch that is vulnerable (98). In patients with significant RV hypertrophy, there may be enlargement of the infundibular (or conal) branch of the RCA. Strictly speaking, this is not anomalous anatomy but, since this branch supplies the musculature of the subpulmonary infundibulum, its prominence may nonetheless challenge the surgeon.
In previous eras, when ventriculotomy and transannular patching were frequent components of TOF repair, the risk of transecting an anomalous coronary vessel was high and patients with abnormal coronary anatomy had increased operative mortality. Preoperative coronary angiography became routine and, if anomalies were suspected, corrective surgery was delayed until midchildhood by use of a palliative shunt procedure (97). Today, there are several surgical techniques for dealing with anomalous coronaries in patients with
TOF and it is rarely necessary to delay surgery, even for abnormalities that might previously have precluded repair. For the majority of patients who have a reasonably sized pulmonary valve annulus, the now standard transatrial–transpulmonary approach (see below) can be used successfully, even in the presence of coronary artery anomalies (99). Several options exist for infants who have both a coronary artery that crosses the RVOT and severe pulmonary annulus hypoplasia. These include RVOT reconstruction with single or separated patches (100,101), placement of an RV-to-PA conduit (100), and use of a native PA flap to protect the anomalous coronary by placing a glutaraldehyde-treated patch between a low ventriculotomy and the PA (102). The technique employed usually depends on surgical preference. It is not always possible to directly identify anomalous coronaries at the time of surgery, so it is important that the coronary anatomy be delineated by careful preoperative echocardiography.
TOF and it is rarely necessary to delay surgery, even for abnormalities that might previously have precluded repair. For the majority of patients who have a reasonably sized pulmonary valve annulus, the now standard transatrial–transpulmonary approach (see below) can be used successfully, even in the presence of coronary artery anomalies (99). Several options exist for infants who have both a coronary artery that crosses the RVOT and severe pulmonary annulus hypoplasia. These include RVOT reconstruction with single or separated patches (100,101), placement of an RV-to-PA conduit (100), and use of a native PA flap to protect the anomalous coronary by placing a glutaraldehyde-treated patch between a low ventriculotomy and the PA (102). The technique employed usually depends on surgical preference. It is not always possible to directly identify anomalous coronaries at the time of surgery, so it is important that the coronary anatomy be delineated by careful preoperative echocardiography.
Aortic Arch
In 20% to 25% of patients with TOF, the aortic arch is right-sided with mirror image branching (i.e., it crosses over the right main bronchus and the first branch is a left-sided brachiocephalic artery, which divides into the left subclavian and left carotid arteries (77,103,104)). In isolation, a right-sided aortic arch in TOF causes no additional symptoms, since it does not produce a complete vascular ring. A right-sided aortic arch may be diagnosed on plain chest x-ray by the absence of the expected left-sided aortic knuckle. By echocardiography, a right aortic arch is best diagnosed from the suprasternal notch. The standard suprasternal view (in which the scanning plane extends from the right nipple to left scapula with the probe’s marker directed toward the patient’s back) will demonstrate only part of a right aortic arch (105). To obtain a full arch image, the transducer must be rotated clockwise from the standard position, so that the marker faces away from the patient and the plane of ultrasound extends from the left border of the sternum to an area just right of the spine (105). From this position, the first branch of a right aortic arch can be traced coursing leftward (in contrast to a left aortic arch where this vessel would course rightward) before bifurcating. A right-sided aortic arch may also be diagnosed by fetal echocardiography, when, in a transverse view, the “sausage-shaped” arch is located to the right of the trachea, rather than its usual left-sided position (106).
Other Associated Cardiac Abnormalities
Important variants of TOF exist which may impact differently on the patient’s physiology, clinical presentation, and surgical management.
Tetralogy of Fallot with Origin of One Pulmonary Artery from the Right Ventricle and One from the Ascending Aorta
In this rare variant, there is discontinuity of the branch pulmonary arteries with one PA (more often the left) originating from the ascending aorta (107,108,109,110,111). Although use of the term is generally discouraged (because it is teleologically incorrect), this anatomy is sometimes known as a “hemitruncus.” It is important to identify patients with this abnormality (which may be overlooked initially) and undertake surgical repair early, to prevent the development of pulmonary arterial hypertension in the left lung.
Occasionally, there may be apparent complete absence of the left PA in the setting of TOF (112,113,114), although such cases (particularly if recognized in early infancy) should be investigated for possible origin of the left PA from a left-sided arterial duct (commonly arising from the base of the left brachiocephalic or subclavian artery), as this can sometimes be probed, dilated, stented, and rehabilitated, even after complete closure of the duct.
 Figure 41.7 MRI of infant with TOF with absent pulmonary valve (A: Right ventricular outflow view and B: Distal pulmonary arteries in sagittal plane). Note the massive dilatation of main and branch pulmonary arteries. RV, right ventricle; PT, pulmonary trunk; LPA, left pulmonary artery; RPA, right pulmonary artery. (Images courtesy of Dr. Shi-Joon Yoo at The Hospital for Sick Children, Toronto.) |
Tetralogy of Fallot with Absence of the Pulmonary Valve (Leaflets)
In approximately 3% to 6% of patients with TOF, the pulmonary valve leaflets are absent (104,115) or only a rudimentary ridge of tissue is present. This variant is associated with significantly aneurysmal main and branch pulmonary arteries that may compromise the airways and respiratory function (Fig. 41.7). The first description
of this anomaly is attributed to Chevers (116) in 1847 and the first detailed case report was published in 1927 by Kurtz et al. (117) who described the clinical features and postmortem findings of an 11-year-old boy. Despite an absence of pulmonary valve tissue, these patients usually exhibit some degree of RVOTO. This is almost always caused predominantly by a ring of tissue present at the level where the pulmonary valve leaflets would be expected, rather than by infundibular stenosis (118,119). An anatomical peculiarity of TOF with absent pulmonary valve is that the arterial duct is nearly always absent (118,120). In about 50% of patients with TOF and absent pulmonary valve, there is also a right-sided aortic arch, and the condition may also be associated with absent or aortic origin of a branch PA.
of this anomaly is attributed to Chevers (116) in 1847 and the first detailed case report was published in 1927 by Kurtz et al. (117) who described the clinical features and postmortem findings of an 11-year-old boy. Despite an absence of pulmonary valve tissue, these patients usually exhibit some degree of RVOTO. This is almost always caused predominantly by a ring of tissue present at the level where the pulmonary valve leaflets would be expected, rather than by infundibular stenosis (118,119). An anatomical peculiarity of TOF with absent pulmonary valve is that the arterial duct is nearly always absent (118,120). In about 50% of patients with TOF and absent pulmonary valve, there is also a right-sided aortic arch, and the condition may also be associated with absent or aortic origin of a branch PA.
Discussion continues as to whether the massive PA dilation already present by the time of birth is primarily attributable to an abnormality of the tissues of the pulmonary vessel, the orientation of the subpulmonary infundibulum, or to severe in utero pulmonary regurgitation (PR) (consequent to absence of the arterial duct and pulmonary valve leaflets) (121). Free PR makes the aneurysmal pulmonary arteries highly pulsatile and they often cause external compression of the airways, sometimes completely obliterating the bronchial lumens during systole. This leads to varying degrees of tracheobronchial malacia and air trapping. Rabinovitch et al. (122) described abnormal branching of the pulmonary arteries with “tufts” of arteries that entwined and compressed the intrapulmonary bronchi on autopsy of two patients with TOF and absent pulmonary valve. The authors suggested that their findings might explain why some patients continue to experience respiratory problems, despite relief of compression of the main stem bronchus by surgical repair (119).
On examination, newborns with TOF and absent pulmonary valve exhibit “to-and-fro” systolic and diastolic murmurs and a single second heart sound, whereas patients with isolated TOF rarely have a diastolic murmur. Most are initially cyanotic, but this usually becomes less apparent during the first week of life (119). Around 40% of patients have some degree of respiratory distress at birth (118). Since there is usually less RVOTO than in other forms of TOF, as pulmonary vascular resistance falls, infants usually develop signs of increased pulmonary blood flow. The chest x-ray of these patients is distinctive and is characterized by a moderately enlarged cardiac silhouette that has a prominent bulge at the upper left cardiac border, caused by the massively dilated proximal pulmonary arteries, and usually normal peripheral vascular markings. Some infants also demonstrate lobar emphysema (123). MRI or CT scanning may add further information about the extent and severity of airway disease (124).
The clinical course and prognosis of patients with TOF and absent pulmonary valve is variable. Although there is probably a spectrum of disease, general consensus divides patients into two groups: those who exhibit severe respiratory problems early in life and those who do not. Patients who present with severe respiratory compromise immediately after birth or in the first weeks of life will generally require urgent intervention and have a worse outcome than those who escape early intervention with relatively minor respiratory involvement. For severely affected infants, some clinical improvement may be gained by prone positioning, which allows the pulmonary arteries to fall forward and away from the bronchi. Otherwise, these patients usually require prompt intubation and positive airway pressure ventilation to maintain their airway. It is clear from early surgical series that infants who present with severe respiratory distress and require preoperative ventilation have the highest surgical mortality (125). However, modern surgical strategies and improvements in intensive care management may have improved outcomes in this group. The authors of a 2007 publication reported successful weaning from ventilation and survival to eventual hospital discharge in all 61 consecutive neonates with TOF and absent pulmonary valve surgically repaired in the preceding 12 years (126).
Several surgical options have been suggested for management of TOF with absent pulmonary valve and, to date, no clear consensus exists as to which is best. Strategies usually focus on intracardiac TOF repair in conjunction with plication and reduction of the pulmonary arteries where necessary. The pulmonary arteries can be reduced in size by removal of tissue from either their anterior or posterior walls. Some surgeons also suspend the left PA to the anterior chest wall on closing the chest, hoping to further release pressure on the bronchi (125,126). An alternative is to perform a variation of the LeCompte maneuver, that is, transect the ascending aorta during repair and move the right PA anterior to it, away from the tracheobronchial tree (127). The surgeon must also decide whether or not to implant a pulmonary valve at the time of the primary procedure, which usually requires placement of a valved RV-to-PA conduit. Some groups also advocate stenting the airway at the time of surgical repair. However, there is no clear consensus about the best approach (128). Even after complete surgical repair with apparent relief of airway obstruction, patients may suffer long-term problems such as recurrent respiratory tract infection, wheezing, and reactive airways disease; some require reintervention for such symptoms (125).
Tetralogy of Fallot with an Atrioventricular Septal Defect
Around 2% of patients with TOF also have an associated atrioventricular septal defect (AVSD) (129). These patients usually present more like patients with isolated TOF than they do like patients with isolated AVSD, since the RVOT limits pulmonary overcirculation and prevents symptoms of heart failure. Furthermore, they will often have left axis deviation on the electrocardiogram. The association should be actively excluded in children with Down syndrome and apparently isolated TOF. It is important to remember in this regard that a primum component may not always be present in such cases, and the presence of an associated AVSD may only be indicated by the presence of a trifoliate (“cleft”) left atrioventricular valve component of a divided common atrioventricular junction (which should be repaired at the time of correction, to avoid later valvar regurgitation). The restriction to pulmonary blood flow protects the pulmonary arteries, and these patients can usually be repaired somewhat later than those with an isolated AVSD. Since surgical repair involves division of the inlet valve as well as closure of the interatrial and interventricular septal defects and reconstruction of the RVOT, this combination of cardiac defects is a particular surgical challenge. Nonetheless, it has been shown that primary repair of both the AVSD and TOF may be performed in the first few months of life and produces better outcomes than an initial palliative approach with full repair delayed until 4 to 6 years of age (129).
Clinical Features and Investigation
Prenatal Diagnosis
Advances in fetal echocardiography, the development of structured screening programs, and increasing sonographer experience have resulted in increased prenatal diagnosis of CHD, including TOF and its variants (130,131). In expert hands, the prenatal diagnosis rate for TOF may be as high as 70% and the diagnostic accuracy can be as high as 90% (130,132). The main source of diagnostic inaccuracy results from diagnosing TOF with pulmonary stenosis in fetuses that are then postnatally found to have TOF with pulmonary atresia (130,132). To some extent, this may be unavoidable since there is good evidence
that the outflow tract obstruction seen in TOF may progress during fetal life and that some fetuses with patent pulmonary valves early in pregnancy will have developed complete pulmonary atresia by the time of birth (87,133). In 2008, Kaguelidou et al. (134) examined outcomes for 238 fetuses diagnosed with either TOF (n = 153) or TOF with pulmonary atresia (n = 65). The median gestational age at diagnosis was 24 weeks, with 45% of cases diagnosed before 24 weeks. Ultrasonography detected coexisting extracardiac anomalies in 70/153 fetuses with TOF. Fetal karyotyping results were available in 132/153 TOF cases and 106/153 fetuses were screened for a 22q11.2 deletion. Of those screened, Trisomy 21 was diagnosed in 4.5%, Trisomy 13 in 0.8%, and other rare (often sex chromosomal) abnormalities in 6%. A 22q11.2 deletion was found in 15% of the 106 fetuses with TOF that were screened. As a result of the antenatal screening, 24% of parents with fetus diagnosed with TOF decided to terminate their pregnancy (a lower proportion than in cases where the diagnosis was TOF with pulmonary atresia). The authors reported that the presence of associated chromosomal abnormalities or severe extracardiac abnormalities were factors that determined parental choice (134).
that the outflow tract obstruction seen in TOF may progress during fetal life and that some fetuses with patent pulmonary valves early in pregnancy will have developed complete pulmonary atresia by the time of birth (87,133). In 2008, Kaguelidou et al. (134) examined outcomes for 238 fetuses diagnosed with either TOF (n = 153) or TOF with pulmonary atresia (n = 65). The median gestational age at diagnosis was 24 weeks, with 45% of cases diagnosed before 24 weeks. Ultrasonography detected coexisting extracardiac anomalies in 70/153 fetuses with TOF. Fetal karyotyping results were available in 132/153 TOF cases and 106/153 fetuses were screened for a 22q11.2 deletion. Of those screened, Trisomy 21 was diagnosed in 4.5%, Trisomy 13 in 0.8%, and other rare (often sex chromosomal) abnormalities in 6%. A 22q11.2 deletion was found in 15% of the 106 fetuses with TOF that were screened. As a result of the antenatal screening, 24% of parents with fetus diagnosed with TOF decided to terminate their pregnancy (a lower proportion than in cases where the diagnosis was TOF with pulmonary atresia). The authors reported that the presence of associated chromosomal abnormalities or severe extracardiac abnormalities were factors that determined parental choice (134).
Postnatal Presentation and Natural History
The clinical signs and symptoms seen in infants born with TOF generally vary in accordance with the degree of RVOTO and frequently the diagnosis will be made following neonatal detection of an asymptomatic murmur. Infants with the most severe obstruction shunt right-to-left across the VSD and have reduced pulmonary flow. They often present with cyanosis in the first days of life. Cyanosis may be recognized at the time of delivery, during routine measurement of newborn oxygen saturation or perhaps only during episodes of crying. Infants with minimal RVOTO usually have a normal, or near normal, oxygen saturation after birth. These babies (“pink tets”) may present with symptoms of cardiac failure (poor feeding, tachypnea, diaphoresis, and failure to thrive) at 4 to 6 weeks of age due to increasing pulmonary flow as their pulmonary vascular resistance drops and they shunt left-to-right across the VSD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


