A basic assumption for the establishment of such a hierarchy assumes that all biological entities have evolved from a single common cellular life-form. Different biological entities have evolved as a result of accumulated changes in DNA that have provided survival advantages in different ecological niches. Species may be classified on the basis of phylogenetic and evolutionary relatedness: members of a given species are the most closely related, different species within a single genus are more closely related to each other than to a species within a different genus, and so on. Newer technologies like microscopy, improved biochemical and physiological analysis, and advanced protein and molecular analytical methods have resulted in an enormous expansion of characteristics that may be studied for the classification of biological entities and validation of taxonomic systems (Woese et al. 1990).
3.2 The Classification and Taxonomy of Viruses
There are a number of excellent texts that discuss the clinical and laboratory aspects of virus biology (Knipe and Howley 2007; Richman et al. 2009; Versalovic 2012). Though viruses are certainly “biological entities,” they are fundamentally different from the cellular life-forms classified by previous taxonomic schemes. Viruses have no autonomous metabolic or replicative ability; they are completely dependent on cellular life-forms. However, within their biological milieu, viruses do replicate and evolve, and they are composed of the same types of organic macromolecules as are cellular life-forms. Because of their intimate relationship with cellular life-forms, it seems legitimate to integrate the schemes for classification of viruses with the schemes used for biological classification of cellular life-forms (Lefkowitz 2012). Initially, various features, like host range, cross-immunity, clinical disease, and pathologic features, were used to classify viruses. Technological advances have led to more detailed and integrated classification, taxonomy, and phylogenetic characterization (evolutionary relatedness) of viruses. Sophisticated nucleic acid sequence analysis has emerged as a powerful tool for virus classification and phylogenetic determination, in spite of some limitations (Holmes 2008; McCormack and Clewley 2002; Zanotto et al. 1996).
A robust system for classification of viruses developed by David Baltimore has gained wide acceptance (Baltimore 1971). Classification is based on the genomic nucleic acid used by the virus (DNA or RNA), strandedness (single or double stranded), and method of replication. The system has been used to define seven classes of viruses:
Class I: Double-stranded DNA (dsDNA)
Class II: Single-stranded DNA (ssDNA)
Class III: Double-stranded RNA (dsRNA)
Class IV: Single-stranded RNA (ssRNA), positive-sense
Class V: Single-stranded RNA, negative-sense
Class VI: Positive-sense ssRNA that replicate by reverse transcription through a DNA intermediate
Class VII: dsDNA viruses that replicate by reverse transcription through an ssRNA intermediate
The International Committee on Taxonomy of Viruses (ICTV) was established by the International Union of Microbiological Societies to oversee and communicate critical issues related to the classification of viruses. This committee was charged with the following:
Developing a nomenclature for specific viral taxa, including virus species
Communicating taxonomic decisions through periodic summary reports, meetings, and journal publications, e.g., Virology Division News in the Archives of Virology
Establishing and maintaining an index of virus names (International Committee on Taxonomy of Viruses 2012)
There are six subcommittees of the ICTV through which proposals related to taxonomy are submitted. These experts evaluate evidence related to proposals for taxonomic changes and make recommendations for final approval by vote of entire ICTV membership. The ICTV recognizes only five of the taxa of classic biological taxonomy:
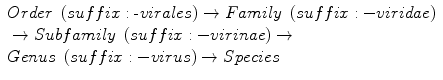
The primary classification of viruses is into species. A virus species is defined as a polythetic class of viruses that constitute a replicating lineage and occupy a specific ecological niche (International Committee on Taxonomy of Viruses 2002). In polythetic classifications, group members share a number of characteristics, but no single characteristic is necessary or sufficient to define members of the group. Higher-level taxa are monothetic, i.e., there are characteristics that are necessary and sufficient to define members of the class. It is important to note that not all viruses can be assigned through all taxonomic levels. Virus species may be assigned to a genus or remain unassigned. Similarly, a genus may be assigned to a family or subfamily, or remain unassigned, and so on up the taxonomic hierarchy. Each genus has a type species. The type species is the virus that necessitated the creation of the genus; it is always linked to the genus.
In the most recent publication (2012), the ICTV recognized 7 orders, 96 families, 22 subfamilies, 420 genera, and 2,618 species. Important characteristics used by the ICTV to define and classify viruses within these taxa include the following:
Susceptible Host Range: Most viruses have a restricted range of hosts which they are able to infect.
Virus Structure: The viral genome is surrounded by a protective shell of proteins called a capsid. The capsid may also enclose proteins, like reverse transcriptase or proteins required for organization of the nucleocapsid. A nucleocapsid refers to a viral nucleus surrounded by an intact capsid. The nucleocapsids of certain viruses are also surrounded by an envelope of host-derived membranes. The complete virus particle is referred to as a virion.
Icosahedral capsids are very common; these quasi-spherical shells are composed of 20 identical equilateral triangles with 30 edges and 12 vertices. Icosahedral capsids are very efficient geometrically (internal volume versus protein content) and genetically (many small sides require fewer and smaller genes to code for capsid proteins). The nucleocapsid proteins of some viruses, like the influenza viruses, form helical tubes with the nucleic acid incorporated directly into the helical structure.
The nucleocapsids of some viruses are surrounded by envelopes composed of lipid bilayers and host- or viral-encoded proteins. Envelopes are typically acquired by budding of the nucleocapsid through a virally modified portion of a specific host-cell membrane (plasma, endoplasmic reticulum, Golgi, nucleus).
The shape of the virus nucleocapsid or intact virion is usually determined by electron microscopy. The shape and dimensions of the nucleocapsid and intact virion, and the presence or absence of an envelope, are useful characteristics for classifying viruses.
Genome: The viral genome is either DNA or RNA; the nucleic acids may be single or double stranded. The genome size may be expressed in terms of kilobases (kb) for single-stranded genomes or kilobase pairs (kbp) for double-stranded genomes. The sequence of genes of positive-sense ssRNA may be directly translated by the host into viral proteins. The sequence of negative-sense ssRNA is complementary to the coding sequence for translation, so mRNA must be synthesized by RNA polymerase, typically carried within the virion, before translation into viral proteins. The sequence of positive-sense ssDNA is the same as that of the mRNA coding for viral proteins; negative-sense ssDNA is complementary to mRNA and may be transcribed into mRNA for viral protein synthesis. Ambisense single-stranded nucleic acids use both positive-sense and negative-sense sequences. The viral nucleic acid may be linear or circular; the nucleic acid may be in the form of a single molecule or broken into two or more segments.
In addition to the type of nucleic acid, the size of the viral genome, measured in number of bases or base pairs, is an important characteristic used for classification.
Nucleic Acid Sequence Analysis: The analysis of specific viral nucleic acid sequences is increasingly used as a powerful tool for taxonomic assignment and assessment of evolutionary relatedness. The utility is greatest for related groups of viruses (Lauber and Gorbalenya2012a, b), but has been challenging for more divergent groups of viruses. Sequence analysis alone has not provided a reliable single criterion on which all viruses may be classified. Construction of a universal phylogenetic tree for viruses, as has been proposed for cellular life-forms, may not be possible for viruses. It is not clear that all viruses emerged from a single progenitor virus; there is evidence for multiple, independent origins of existing viruses. Phylogenetic analysis using nucleic acid sequences is further complicated by recombination, reassortment, incorporation of host nucleic acid sequences, and other factors (Domingo 2007; Holmes 2011).
Currently, expert consensus, considering laboratory, phenotypic, clinical, and other characteristics, remains the most accurate and robust method for the classification and taxonomic assignment of viruses. Note that the formal names assigned at all taxonomic levels are italicized, while the common names, which are often used clinically, are not italicized. The viruses that have been associated with human infections are shown in Table 3.1.
Table 3.1
Virus families associated with human infections
Nucleic acid | Family | Segments | Genome | Nucleocapsid | Envelope |
|---|---|---|---|---|---|
Class I: dsDNA | Adenoviridae | Unsegmented | Linear, 30–38 kbp | Icosahedral | No |
Herpesviridae | Unsegmented | Linear, 125–240 kbp | Icosahedral | Yes | |
Papillomaviridae | Unsegmented | Linear, 7–8 kbp | Icosahedral | No | |
Polyomaviridae | Unsegmented | Circular, 5 kbp | Icosahedral | No | |
Poxviridae | Unsegmented | Linear, 130–375 kbp | Ovoid | Yes | |
Class II: ssDNA | Anelloviridae | Unsegmented | Circular, 3–4 kb. Negative-sense | Icosahedral | No |
Parvoviridae | Unsegmented | Linear, 4–6 kb. Ambisense | Icosahedral | No | |
Class III: dsRNA | Reoviridae | 10–12 segments | Linear, 19–32 kbp | Icosahedral | No |
Class IV: ssRNA, positive-sense | Astroviridae | Unsegmented | Linear, 6–7 kb | Icosahedral | No |
Caliciviridae | Unsegmented | Linear, 7–8 kb | Icosahedral | No | |
Coronaviridae | Unsegmented | Linear, 27–32 kb | Helical | Yes | |
Flaviviridae | Unsegmented | Linear, 10–12 kb | Spherical | Yes | |
Hepeviridae | Unsegmented | Linear, 7 kb | Icosahedral | No | |
Picornaviridae | Unsegmented | Linear, 7–9 kb | Icosahedral | No | |
Togaviridae | Unsegmented | Linear, 10–12 kb | Icosahedral | Yes | |
Class V: ssRNA, negative-sense | Arenaviridae | 2 segments | Linear, 11 kb. Ambisense | Sphere | Yes |
Bornaviridae | Unsegmented | Linear, 9 kb | Not defined | Yes | |
Bunyaviridae | 3 segments | Linear, 11–19 kb. Ambisense | Helical | Yes | |
Filoviridae | Unsegmented | Linear, 19 kb | Helical | Yes | |
Orthomyxoviridae | 6–8 segments | Linear, 10–15 kb | Helical | Yes | |
Paramyxoviridae | Unsegmented | Linear, 13–18 kb | Helical | Yes | |
Rhabdoviridae | Unsegmented | Linear, 11–15 kb | Bullet shaped | Yes | |
Deltavirus | Unsegmented | Circular, 2 kb | Spherical | Yes | |
Class VI: ssRNA, positive-sense, reverse transcribed | Retroviridae | Unsegmented, diploid | Linear, 7–13 kb | Icosahedral (spherical or cone-shaped core) | Yes |
Class VII: dsDNA, reverse transcribed | Hepadnaviridae | Unsegmented | Circular, partially dsDNA, 3kbp | Icosahedral | Yes |
3.3 Viruses and the Lung
Among the families of viruses able to infect humans and other vertebrate hosts, there are many species that target and cause disease in the lung. These viruses commonly use airborne transmission as an effective mode of transmission between an infected host and a new susceptible host. Characteristics of viruses that directly or indirectly cause pulmonary disease are discussed in this section.
Adenoviridae: Adenoviruses are pathogenic for humans and other vertebrate species. A structural protein at each of the 12 of the icosahedral nucleocapsid vertices anchors a rodlike projection with a terminal knob, which interacts with specific host surface receptor molecules and which confers the hemagglutination pattern and tissue tropism for the different groups of adenoviruses. The genome encodes ~40 genes (Davison et al. 2003a), including common genes and species-specific genes. Genes are grouped into early, delayed early, and late transcribed genes. The genome contains inverted repeat sequences at both ends. Sequences of both DNA strands are transcribed to mRNA; mRNA splicing is used for expression of many adenovirus genes.
The family Adenoviridae has not been assigned to an order. Within this family, there are five genera. The seven species that cause human infection are Human adenovirus A, B, C, D, E, F, and G, all within the Mastadenovirus genus; there are 57 accepted serotypes (Buckwalter et al. 2012). Endemic respiratory infections are most commonly caused by serotypes of Human adenovirus C (the type species of the genus); most epidemic respiratory infections are caused by serotypes within species adenovirus B and adenovirus E.
Arenaviridae: Arenaviruses may cause several hemorrhagic fever syndromes. Specific rodents are the reservoir for each arenavirus; human disease is incidental and is usually transmitted by infectious aerosols. Viruses of this family are enveloped; evenly spaced glycoprotein complexes (a tetramer of viral GP2 with viral GP1 ionically bound as a globular head) are attached to the envelope giving complete virions a studded spherical morphology. Complete virions are ~100 nm in diameter, but show significant pleomorphism (range, 60–300 nm). The genome is divided into two segments which are complexed with nucleoproteins (Peters 2009). Complementary sequences at the 3′ and 5′ ends of each segment result in the formation of two circular nucleocapsids. Arenaviruses use both negative-sense and ambisense coding strategies. Host ribosomes are often incorporated within the envelope of complete virions.
This family of viruses is not assigned to an order. There is one genus, Arenavirus, with 25 species that fall into two complexes on the basis of serologic and genetic relatedness. The Old World, or African, species include Lassa virus (Lassa fever) and Lujo virus. The New World species include Guanarito virus (Venezuelan HF), Junín virus (Argentine HF), and Machupo virus (Bolivian HF). The type species of the genus Arenavirus is lymphocytic choriomeningitis virus.
Bunyaviridae: Bunyaviruses may cause several hemorrhagic fever syndromes. Viruses within this family are enveloped with protein complexes (heterodimers of glycoproteins Gn and Gc) anchored to the lipid bilayer. Complete virions are spherical (80–120 nm diameter) with projecting spikes; the pattern of spikes varies among different species. The genome consists of three ssRNA strands, designated short (1–2.2 kb), medium (3.5–6 kb), and long (6.3–12 kb) (Mertz 2009). The RNA is complexed with nucleocapsid protein to form three helical nucleocapsids (small, medium, and large) within the envelope. Some bunyaviruses use negative-sense coding exclusively; some use a combination of negative-sense and ambisense coding.
The family Bunyaviridae is not assigned to an order. There are five genera in this family. There are 24 species in the genus Hantavirus, including Andes virus, Hantaan virus (the type species), Puumala virus, Seoul virus, and Sin Nombre virus. Rodents, not arthropods, are the reservoir for species of the genus Hantavirus. In the genus Nairovirus, there are seven species, including Crimean-Congo hemorrhagic fever virus (tick vector). In the genus Phlebovirus, there are nine species, including Rift Valley fever virus (the type species, mosquito vector) and Sandfly fever Naples virus (sandfly vector).
Coronaviridae: Transmembrane proteins produce blunt projections from the surface of coronaviruses, resulting in a “crown-like” appearance on electron microscopic studies (100–160 nm in diameter). Translation of the coronavirus genome is unique and includes production of polyproteins, discontinuous synthesis, overlapping reading frames, ribosomal frame shifting, and post-translational proteolytic processing (Marra et al. 2003; Rota et al. 2003; Theil et al. 2003). The major structural proteins, spike glycoprotein (S), membrane glycoprotein (M), nucleocapsid phosphoprotein (N), hemagglutinin-esterase glycoprotein (HE), and envelope protein (E), are present in all coronaviruses. Nonstructural proteins are encoded in 5–10 unique or overlapping reading frames (Lai et al. 2007).
The human coronaviruses are assigned to the order Nidovirales, family Coronaviridae, and subfamily Coronavirinae. There are four genera and three serological groups. Relevant viruses include Human coronavirus 229E and Human coronavirus NL63 of the genus Alphacoronavirus (antigenic group I), Human coronavirus HKU1, Betacoronavirus 1 and Severe acute respiratory syndrome-related coronavirus of the genus Betacoronavirus (antigenic group II).
Filoviridae: Filoviruses may cause several hemorrhagic fever syndromes. The filoviruses have a unique threadlike morphology. The helical nucleocapsids are surrounded by an envelope studded by spikes formed by a single type of glycoprotein (GP). The genome consists of a single segment of negative-sense ssRNA that encodes for seven proteins (Kuhn et al. 2010). The presence of gene overlap for several genes is an unusual feature of filoviruses. In ebolaviruses, the surface glycoprotein is encoded by two adjacent reading frames. A truncated version (sGP), which lacks the hydrophobic anchor, results from translation of the upstream reading frame only. This protein is secreted from cells and may serve as a decoy for the host’s immunological response. The full-length GP is formed only when the RNA polymerase misreads a poly-U editing site between the reading frames. The full-length GP is inserted, as homotrimers, into the host membranes that will form the virion envelope. A helical nucleocapsid is formed by association of the ssRNA with nucleoproteins. The nucleocapsid is ~50 nm in diameter, with a central axial space ~20 nm in diameter. The nucleocapsid is attached to the envelope by matrix protein. The complete virions are ~80 nm in diameter, but the virion length may vary from 800 to 10,000 nm.
The family Filoviridae is assigned to the order Mononegavirales. There are two genera within the family Ebolavirus and Marburgvirus. There are five ebolavirus species, including Sudan ebolavirus and Zaire ebolavirus (the type species). The genus Marburgvirus consists of one species, Marburg marburgvirus. Humans and nonhuman primates are susceptible to ebolavirus and marburgvirus infection; the host reservoirs for these viruses are unknown. Humans may be infected sporadically by presumed contact with the host species or by direct contact with virus containing body fluids taken from acutely infected humans or nonhuman primates. Nosocomial and laboratory-acquired infections are well described.
Flaviviridae: Flaviviruses may cause several hemorrhagic fever syndromes. Hepatitis C virus is also a flavivirus species. Flaviviruses are surrounded by an envelope studded with dimers of viral E glycoprotein and M protein which give the mature virion a herringbone appearance with icosahedral symmetry. The genome consists of a single segment of positive-sense ssRNA (Chambers et al. 1990; Osatomi and Sumiyoshi 1990). Cyclization of the genome, through hybridization of RNA sequences of the 5′ and 3′ ends of the genome, may be required for mRNA synthesis (Alvarez et al. 2005). There is a long open reading frame that codes for three structural proteins at the 5′ end; downstream of this region are genes for seven nonstructural proteins (Thurner et al. 2004). The positive-sense genome is directly translated into a large polyprotein, which undergoes intra- and post-translational cleavage. Strain evolution and clinical diversity have been driven by a high rate of mutation at replication and through molecular recombination. The nucleocapsid is formed by interaction of genomic RNA with capsid proteins. The complete virion has a spherical morphology approximately 50 nm in diameter.
This family of viruses is not assigned to an order. There are four genera within the family Flaviviridae. Within the genus Flavivirus, there are 53 species, including Dengue virus (Simmons et al. 2012), Kyasanur Forest disease virus, Omsk hemorrhagic fever virus, and Yellow fever virus (the type species). In addition to viruses that cause hemorrhagic fever syndromes, the family Flaviviridae includes many species of neurotropic viruses that cause encephalitis and other CNS infections, like Japanese encephalitis virus, St. Louis encephalitis virus, Tick-borne encephalitis virus, and West Nile virus. Most human flavivirus infections are transmitted by mosquitoes or ticks.
Hepatitis C virus (HCV) is the type species of the genus Hepacivirus in the family Flaviviridae. The physical properties of HCV have not been as well defined as other flaviviruses because there is no efficient method for in vitro replication of HCV. Virion morphology is consistent with other flaviviruses; complete, enveloped virions have a diameter of 55–65 nm. The single segment positive-sense ssRNA is ~9.6 kb in length (Hijikata et al. 1991). A single open reading frame is flanked by highly conserved regions at the 5′ and 3′ ends. Cap-independent protein synthesis, typical of Flavivirus species, is initiated at an internal ribosomal entry site (IRES) within the 5′ untranslated region. This results in synthesis of a polyprotein that undergoes cleavage and further processing during and after translation. A unique and highly conserved sequence upstream of the IRES interacts with liver-specific microRNA and is required for efficient replication. Circulating HCV is associated with host LDL/VLDL, which may play a role in delivery of virions to hepatocytes.
The error-prone RNA polymerase and high replication rate of HCV has resulted in a great genetic diversity and heterogeneity of clinical isolates. HCV isolates can be grouped by genotypic analysis into six groups and many subgroups. There are differences with respect to responses to antiviral therapy among the genotypes, but intrinsic virulence is similar. The vast majority of strains in the United States are genotypes 1a, 1b, and 2, whereas Central African strains are almost exclusively genotype 4.
Hemorrhagic Fever (HF) Syndromes: Viral hemorrhagic fever syndromes may be caused by many species of viruses from four different families: Arenaviridae, Bunyaviridae, Flaviviridae and Filoviridae; all are single-stranded RNA viruses. See the discussions above for specific information related to these virus families.
Typical symptoms of viral hemorrhagic fever infection include fever, malaise, hypotension, and coagulation defects. With the exception of dengue, the other HF viral agents are maintained in nonhuman vertebrate hosts; humans are coincidental, dead-end hosts. In dengue, human infection is maintained through a mosquito vector. The epidemiologic distribution of disease reflects the geographic range of the reservoir host.
HF viruses primarily infect dendritic cells, macrocytes, and monocytes, which are present in virtually all tissues and organ systems; parenchymal cells may also be susceptible to infection, depending on the virus. Infected cells release mediators that result in marked increased vascular permeability, compromising the function of critical organ systems. Suppression of cellular type 1 interferon response is a significant contributor to pathogenesis (Habjan et al. 2008).
Hepadnaviridae: In the family Hepadnaviridae, there are two genera, Avihepadnavirus (two species) and Orthohepadnavirus (four species); hepatitis B virus (HBV), the type species of Orthohepadnavirus, is only human pathogen in family. The family Hepadnaviridae is not assigned to an order. Eight distinct HBV genotypes (A–H) and subtypes can be recognized on the basis of antigenic or sequence variation. The genotypes show geographic and ethnic variability; the HBV genotype influences the severity and outcome of disease (Garfein et al. 2004; Lin and Kao 2008).
The complete, enveloped HBV virion (Dane particle) is 42–47 nm in diameter. The icosahedral nucleocapsid (~28 nm in diameter) of the virion contains a single molecule of partially double-stranded DNA with a DNA-dependent polymerase covalently linked to the 5′ end of the complete DNA strand, hepatitis B e antigen (HBeAg) and hepatitis B core antigen (HBcAg). The nucleocapsid is surrounded by an envelope derived from host-cell membrane and viral envelope proteins, including hepatitis B surface antigen. The genome of HBV is a circular, partially double-stranded DNA molecule which is replicated by a unique process of reverse transcription of an RNA intermediate. The minus DNA strand runs the entire length of the HBV genome; the plus strand covers only about two-thirds of the genome. The genome is replicated by synthesis of a full-length ssRNA transcript (pre-genomic RNA), followed by dsDNA synthesis by reverse transcription of the ssRNA by viral-encoded reverse transcriptase/DNA polymerase. All viral proteins are also transcribed from the minus DNA strand. There are four overlapping open reading frames, all read in the same direction (Liang 2009).
Herpesviridae: The herpesvirus species associated with human infections (HSV-1, HSV-2, CMV, EBV, VZV, HHV-6, HHV-7, and HHV-8) belong to the family Herpesviridae within the order Herpesvirales. There are four subfamilies of the Herpesviridae: Alphaherpesvirinae (5 genera), Betaherpesvirinae (4 genera), Gammaherpesvirinae (4 genera), and a single genus in an unassigned subfamily. Specific human herpesviruses are discussed in the sections below.
The herpesviruses are double-stranded DNA viruses. The icosahedral capsid (~100 nm diameter) is surrounded by an envelope studded by a variety of short glycoproteins. The nucleocapsid is a dense toroid complex with an outer diameter ~70 nm and inner diameter ~18 nm. An irregular “tegument” fills the space between the envelope and capsid. Depending of the thickness of the tegument layer, complete virions range in size from ~125 to >250 nm. The size and organization of the dsDNA genome varies among the species causing human disease (McGeoch et al. 2006). The genomes of human herpesviruses include unique sequences and repeated sequences. Though the genomes are linear in virions, they circularize in the nucleus of infected cells, which is mediated through repeat sequences at both ends of the dsDNA genome. For HHV6 and HHV7 (class A genome), a large unique sequence region is flanked by a region that is repeated at both ends of the linear strand of dsDNA. The genome of EBV and the Kaposi’s sarcoma-associated herpesvirus (class C genome) have smaller left and right terminal repeat sequences, while repeat sequences R1 to R4 divide the unique sequence nucleic acid into four discrete regions. For VZV (class D genome), a large terminal sequence is inverted and inserted into the genome, resulting in a large unique sequence region (UL) and a small unique sequence region (US). HSV-1, HSV-2, and CMV (class E genomes) are the most complex. There are repeat sequence regions at both ends of the linear dsDNA molecule. The unique sequence dsDNA is divided into UL and US regions by a sequence composed of juxtaposed copies of the terminal repeat sequences inserted in an inverted orientation.
Typical of dsDNA viruses, a large number of proteins are produced by various herpesviruses. The organization of the coding regions is complex, with 3′ and 5′ reading frames, gene overlap, spliced genes, and intron regions. Forty genes are conserved among the α-, β-, and γ-herpesviruses. These core genes are divided among seven gene blocks (Albà et al. 2001); within each block the order and polarity of genes are conserved, including genes for gene regulation, nucleotide metabolism, DNA replication, virion maturation, envelope glycoprotein synthesis, and capsid, fusion and tegument protein synthesis.
Diseases caused by human herpesviruses range from systemic to localized infection of virtually all organ systems, although the host-cell range and typical disease characteristics vary by species. A characteristic of herpesvirus infections is latency, which is commonly associated with reactivation and symptomatic infections (e.g., shingles). While active infection with herpesviruses results in the destruction of the infected host cell, latently infected cells remain viable. In latently infected cells, the viral genome forms circularized molecules within the host nucleus with limited expression of viral genes.
Cytomegalovirus: Human cytomegalovirus (hCMV) is the most complex human herpesvirus. The complete virions of human cytomegalovirus range in diameter from ~200 to 300 nm. The Golgi-derived envelope is studded with 20 or more virally encoded glycoproteins. The icosahedral nucleocapsid (~125 nm) includes five capsid proteins enclosing a class E genome (~230 kbp linear dsDNA) (Davison et al. 2003b; Dunn et al. 2003), as described above. The hCMV tegument is composed of at least 27 virally encoded proteins and other viral and host-cell macromolecules.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


