(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
While progress achieved in mitochondrial research has been mainly used in the discovery of the pathogenesis of neuromuscular, cardiovascular, and other diseases, more specific studies are needed to determine the severity of heart mitochondrial defects (either primary or secondary to myocardial alterations) and its contribution to the pathophysiology of cardiovascular diseases. This new knowledge could be applied to identify specific mitochondrial-targeted drugs to be used not only in the heart failure syndrome but also to reverse, or at least to slow, potential changes in cardiac cell structure and function which occur in many other cardiovascular diseases as well as in the aging heart.
Introduction
Mitochondria play a central role in both the maintenance of normal cardiac function and cardiac pathophysiology. However, many fundamental questions remain yet to be answered concerning the molecular mechanisms underlying this role. Understanding of these mechanisms allows this knowledge to be applied to clinical diagnosis and treatment of mitochondrial-based cardiac diseases. To address this critical issue, new technologies have been developed; some of them have been tested in animal models only, while others are currently being investigated in clinical trials. Despite the enormous progress in our understanding of the pathogenesis of mitochondrial diseases, no efficient and reliable therapies for mitochondrial disorders have yet been designed. Although several pharmacological treatments have been proposed, there is no strong evidence that these therapeutic approaches are effective. For example, compounds, such as vitamins and metabolic cofactors (e.g., riboflavin, thiamine, tocopherol [vitamin E], folic acid, succinate, ascorbate [vitamin C], menadione, L-carnitine, and CoQ10), have been used in different combinations as therapeutic “cocktail.” While beneficial effects have been reported in several patients with cardiomyopathy, no large-scale, rigorously controlled studies have been carried out to support their efficacy. New delivery systems/drug carriers of mitochondrial-specific compounds also have been currently tested, some with promising results. Other approaches including the use of stem cell (SC) transplantation and gene therapy for mitochondrial-based diseases have also been tested. In this chapter, available and forthcoming methodologies will be discussed.
Treatment of Mitochondrial Respiratory and Metabolic Defects
Despite great progress in our understanding of the pathogenesis of mitochondrial-based cardiac diseases, therapeutic interventions for their effective treatment are still lagging behind. While various pharmacological treatments have recently been suggested, no wide-accepted consensus on their efficacy as well as their possible side effects has yet been reached [1]. Mainly a number of vitamins and metabolic cofactors (e.g., riboflavin, thiamine, tocopherol, folic acid, succinate, ascorbate, menadione, L-carnitine, and CoQ10) have been employed, usually in various combinations as a therapeutic “cocktail.” These compounds may affect mitochondrial physiology at different levels, including bypassing potential blocks in the respiratory chain caused by defects in specific electron transport chain (ETC) complexes, facilitating scavenging of reactive oxygen species (ROS) and metabolites, which might be reduced due to either oxidative damage, defective synthesis, or transport-caused mitochondrial dysfunction.
Definitive diagnosis of the mitochondrial disorder, i.e., identification of the specific disease-causing molecular defects responsible, is critical to define the appropriate treatment. For instance, treatment with the appropriate compound in patients with either primary carnitine or CoQ10 deficiency has shown to be unambiguously (mainly in cases of carnitine deficiency) lifesaving. The effectiveness of carnitine in children with carnitine-deficiency cardiomyopathy underscores the importance of evaluating carnitine levels in children with clinically unexplained cardiomyopathy, as well as the importance of timing of such treatment [2, 3]. In addition, patients with cardiomyopathy and cytochrome c oxidase (COX) deficiency due to mutations in SCO2, a COX-assembly protein implicated in the incorporation of copper into the COX holoenzyme, have shown a positive response to copper supplementation [4]. However, in many cases of mitochondrial cardiomyopathy (MCM), the precise molecular basis of the mitochondrial defect is not known; therefore, the use of such generic “cocktails” may not be effective.
Severe defects in the respiratory chain may result in accumulation of upstream metabolites including pyruvate, lactate, and the transaminated product, alanine. Levels of all three compounds are elevated in blood and urine of patients with mitochondrial myopathies. Lactic acidosis has a spectrum of neurotoxic effects and may be effectively controlled by the use of dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase (PDH) kinase. Also, improvement has been noted in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) syndrome treated with DCA [5]. By rapidly stimulating PDH activity, and promoting aerobic glucose oxidation in myocardial cells, DCA can improve myocardial function in conditions with limited oxygen availability (e.g., ischemia/reperfusion and congestive heart failure) preventing mitochondrial energy failure associated with these states [6]. In addition to decreasing lactic acidosis, the use of DCA has been associated with suppression of myocardial long-chain fatty acid (FA) metabolism and increased left ventricular stroke work and cardiac output, without changes in myocardial oxygen consumption. Moreover, DCA-dependent stimulation of PDH activity, which is otherwise depressed in the diabetic heart, can be beneficial. DCA-mediated increase in glucose metabolism rate, in combination with the use of partial FA oxidation (FAO) inhibitors, has been considered a potential therapeutic approach to diabetic cardiomyopathy [7].
In patients with COX deficiency, DCA in combination with aerobic training may improve exercise capacity and aerobic metabolism [8]. Albeit aerobic exercise in patients with mitochondrial diseases has shown beneficial effects, such as increasing work and oxidative capacity, exercise tolerance and aerobic training are not systematically evaluated in patients with MCM.
Treatment with Antioxidants
Mitochondria are an important source of oxygen free radicals, and increased ROS generation plays an important role in the pathogenesis of mitochondrial-based cardiac disorders, which suggest that antioxidants may have a beneficial effect in these disorders [9, 10]. As noted previously, mitochondrial ROS and oxidative stress (OS) are implicated in the pathogenesis of several conditions, including the cardiomyopathy induced by doxorubicin toxicity, the cardiomyopathy associated with Friedreich ataxia (FRDA), cardiac damage induced by ischemia/reperfusion [10, 11], and the cardiomyopathy associated with aging, which is mediated by accumulation of mitochondrial DNA damage secondary to mitochondrial OS [10–12]. Tsutsui et al. [13] have observed that chronic increases in mitochondrial ROS output can lead to a destructive cycle of mitochondrial DNA (mtDNA) as well as a mitochondrial functional decline, further increasing ROS generation, and cellular injury. Moreover, ROS induce myocyte hypertrophy, apoptosis, and interstitial fibrosis by activating matrix metalloproteinases. These cellular events appear to play a significant role in the development and progression of maladaptive myocardial remodeling and failure. Hence, OS and mtDNA defects appear to be important therapeutic targets. The investigators have also noted that overexpression of peroxiredoxin-3 (Prx-3), a mitochondrial antioxidant member of the family of thioredoxin peroxidases that uses mitochondrial thioredoxin-2 (Trx2) as a source of reducing equivalents to scavenge hydrogen peroxide (H2O2), or mitochondrial transcription factor A (TFAM), could ameliorate the decline in mtDNA copy number observed in heart failure (HF) (Fig. 23.1). Importantly, Prx-3 overexpression can also prevent decrease in mitochondrial function. Thus, the activation of Prx-3 or TFAM gene expression can ameliorate the pathophysiological processes seen in mitochondrial dysfunction and myocardial remodeling, and inhibition of OS and mtDNA damage might be a novel and effective treatment strategy in HF cases.
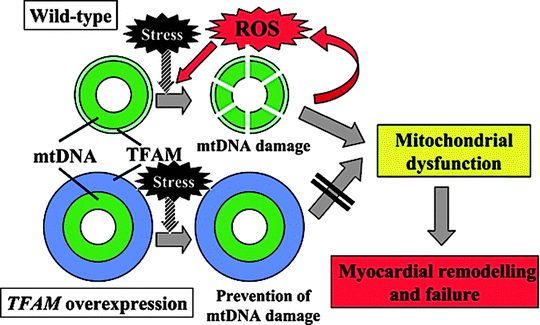
Fig. 23.1
Proposed mechanisms for overexpression of mitochondrial transcription factor A (TFAM) gene to prevent mitochondrial DNA (mtDNA) damage, oxidative stress, and myocardial remodeling and failure (Adapted from Tsutsui et al. [13] with permission from Elsevier)
Besides Prx-3, other antioxidative compounds including CoQ10, vitamin E, dexrazoxane, and idebenone have been used [13–16] Table 23.1. Again, left ventricular remodeling and HF after myocardial infarction in mice could be prevented by overexpression of Prx-3 [17]. Whereas the beneficial effect appears to be related to the scavenging of mitochondrial ROS in cardiomyocytes [18], which is supported by the observation that cardiomyocyte-specific knockouts of MnSOD develop HF [19], the demonstration that overexpression of endothelial mitochondrial Trx2 protects from atherosclerosis [20] strongly suggests that endothelial mitochondria may be a valid therapeutic target [18]. In doxorubicin-induced cardiomyopathy, the free-radical scavenger dexrazoxane has been shown to protect the heart from doxorubicin-associated oxidative damage; its use has been recommended in children with acute lymphoblastic leukemia treated with doxorubicin chemotherapy to decrease the myocardial damage [21]. Of interest, data collected from mice showed that long-term oral intake of nitrate attenuates doxorubicin-induced ventricular dysfunction, cell death, OS, and mitochondrial respiratory chain damage suggesting that nitrate may be a promising therapeutic agent against doxorubicin-induced cardiotoxicity [22].
Using transgenic (Tr) mice with cardiomyocyte-specific overexpression of human cytochrome P450 (CYP)2J2, Zhang et al. [23] have studied how to prevent the toxicity resulting from acute or chronic doxorubicin (Dox) administration. Acute treatment has resulted in marked increases in serum lactate dehydrogenase and creatine kinase levels, which were significantly greater in wild-type (WT) than in CYP2J2 Tr mice. Acute treatment has also resulted in less activation of stress response enzymes in CYP2J2 Tr mice. In addition, CYP2J2 Tr hearts have exhibited less Dox-induced cardiomyocytes apoptosis compared to WT hearts. After chronic Dox treatment, comparable decreases in body weight have been observed in WT and CYP2J2 Tr mice. However, cardiac function, determined using left ventricle shortening fraction with M-mode echocardiography, has been significantly higher in CYP2J2 Tr than in WT hearts. WT mice have also had larger increases in β-myosin heavy chain and cardiac ankryin repeat protein compared to CYP2J2 Tr mice. CYP2J2 Tr hearts had a significantly higher rate of Dox metabolism than WT hearts. Moreover, in vitro findings from H9c2 cells demonstrated that epoxyeicosatrienoic acids (EETs) attenuated Dox-induced mitochondrial damage. Taken together, these observations suggest that cardiac-specific overexpression of CYP2J2 contained Dox-induced toxicity.
Information gathered from several studies has shown that both CoQ10 and idebenone markedly improved cardiac function and reduced myocardial hypertrophy in patients with Friedreich ataxia (FRDA) [24–26], a disease that results from increased/expansion in copies of the GAA trinucleotide repeat in the frataxin gene. Also, idebenone seems to ameliorate the cardiac dysfunction reported in MCM [14]. Nonetheless, albeit this antioxidant protein has a rapid and dramatic effect in most patients with MCM, in some cases the response to idebenone is variable. It is worth noting that idebenone may have a rather wide spectrum of actions with neurological improvement in FRDA, but without noticeable effect on ataxia [27]. Parenthetically, while a previous unifying hypothesis suggested that frataxin deficiency leads to a vicious circle of faulty iron handling, impaired iron-sulfur cluster synthesis, and increased oxygen radical production, newly revised data from cell and animal models suggest that the iron accumulation is inconsistent and rather a late event in FRDA, where frataxin deficiency does not always impair the activity of iron-sulfur cluster-containing proteins [28]. Probably, frataxin deficiency is mainly associated with increased sensitivity to ROS rather than with an increased in ROS production. Altogether, data obtained from experimental research and clinical observations suggest that the first consequence of frataxin depletion is an abnormal oxidative status, which then initiates the pathogenic mechanism underlying FRDA [28].
Besides its role as an antioxidant, CoQ10 serves multiple cellular functions including participation as an electron carrier in the respiratory chain and as an activating cofactor for the mitochondrial uncoupling proteins. Moreover, it has been found that CoQ10 has beneficial effects in several neurological disorders associated with cardiac dysfunction or conditions including MELAS and Kearns-Sayre syndrome (KSS) syndromes [5] [5]. Treatment with CoQ10 at relatively high doses (range 60–150 mg/day) results in significant reduction in the cardiac conduction abnormalities observed in patients with KSS or chronic progressive external ophthalmoplegia (CPEO) syndromes [20, 29]. Clinical improvement has been also reported in patients with congestive heart failure (CHF) after CoQ10 supplementation to standard medical therapy [30]. However, the size of the sample and the design used have raised concern as to the validity of a systematic clinical use of CoQ10 in CHF; a large double-blind, multisite clinical trial appears to be called for to probe the efficacy of this compound [31, 32].
Primary CoQ10 deficiencies have been found in mitochondrial myopathies with involvement of various genes of the CoQ10 biosynthetic pathway, and in some cases the response to CoQ10 treatment was positive. Interestingly, the experience learned on the effect of CoQ10 therapy in Parkinson’s and Huntington’s diseases has been also applied to patients with FRDA, in whom CoQ10 and other quinones have been tested [33].
Antioxidant Defense in Heart Failure/ROS Regulation of Signaling Pathways
It is important to emphasize that ROS and cellular redox states regulate an extensive number of vital pathways in the myocardium, including energy metabolism, survival and stress responses, apoptosis, inflammatory response, and oxygen sensing (Fig. 23.2).
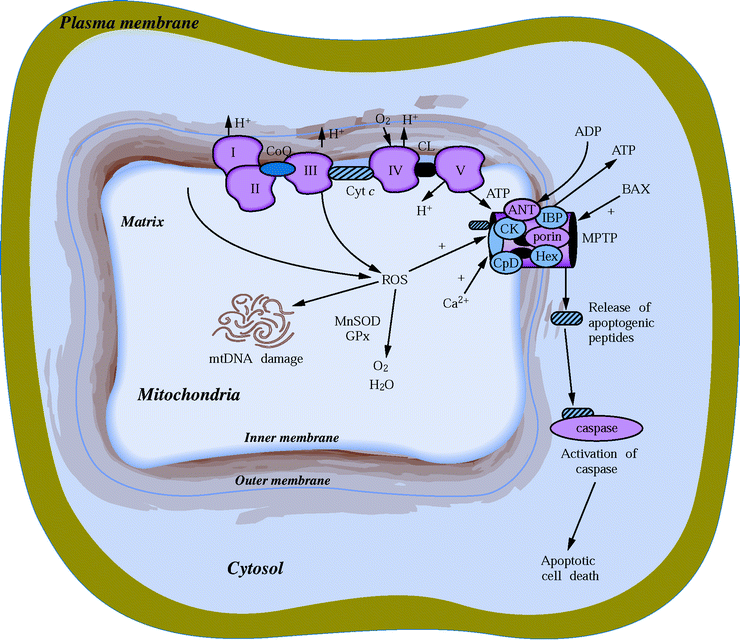
Fig. 23.3
Cellular ROS generation and metabolism. Sites of mitochondrial superoxide O2•− radical (via respiratory complexes I, II, and III) and cytosolic O2•− generation (by NADPH oxidase or xanthine oxidase) are depicted. Also shown are reactions of the O2•− radical with NO to form the highly reactive peroxynitrite, which can target MPTP opening and the inactivation of mitochondrial aconitase by O2•−. MnSOD (in mitochondria) and CuSOD (in cytosol) to form H2O2 are also displayed. The H2O2 is then either further neutralized in the mitochondria by glutathione peroxidase (GPX) and glutathione, in the peroxisome by catalase, or in the presence of Fe2+ via the Fenton reaction, which forms the highly reactive •OH radical that can cause severe lipid peroxidation and extensive oxidative damage to proteins and mtDNA
While ROS play a central role in ischemia and reperfusion injury, the response to antioxidant therapy varies significantly depending on whether animal models or isolated cell models have been used. This may explain at least in part why in clinical trials antioxidant therapies have shown mixed results (Fig. 23.3).
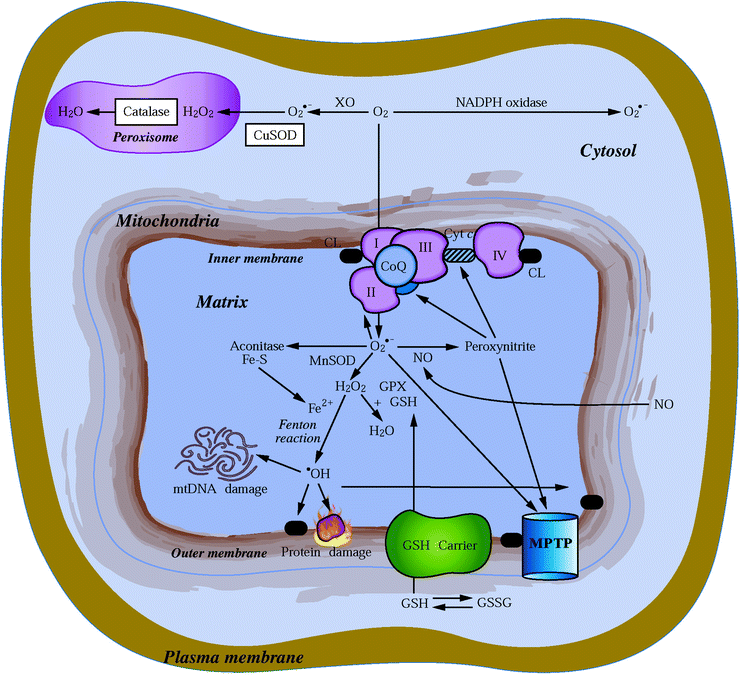
Fig. 23.2
ROS generation by mitochondria produces mtDNA damage and impacts the expression of genes encoding key elements of the apoptotic pathway
Powerful cell-damaging ROS can be neutralized by an array of protective antioxidant scavenger enzymes as well as by various lipid and water-soluble compounds including ascorbic acid, glutathione, thioredoxin, and α-tocopherol. The antioxidant enzymes are located in several cellular compartments including the mitochondria (e.g., MnSOD, glutathione peroxidase (GPx), thioredoxin reductase), peroxisomes (e.g., catalase), microsomes (e.g., cytochrome P450 [CYP450]), and the cytosol (e.g., CuSOD and cytosolic thioredoxin reductase). While levels of myocardial mitochondria antioxidant enzymes are lower than in liver mitochondria, the antioxidant capacity of the heart is generally sufficient to handle normal levels of ROS; however, it is insufficient to deal with the greater ROS accumulation that occurs in myocardial ischemia [34].
A mitochondrial isoform of catalase with low specific activity has been found in rats [35, 36]. This mitochondrial catalase activity has been detected in the heart but not in liver or skeletal muscle, and it appears to be upregulated during caloric restricted (CR) diets and in the diabetic heart [37–39]. Catalase participates in the prevention of excess lipid peroxidation in myocardial ischemia [40], although a mitochondrial-specific catalase has not been found in the heart of transgenic mice even after overexpression of the catalase gene [41].
While superoxide dismutases (SOD) catalyze the removal of superoxide radicals by the formation of H2O2, GPx catalyzes the breakdown of H2O2 to water and oxidized glutathione (GSSG) by using reduced glutathione (GSH) as depicted in Fig. 23.3. Since GPx is located in both the mitochondria and cytosol, H2O2 can be removed from either compartment depending on the availability of glutathione. A small fraction of the total cellular pool of GSH is sequestered in mitochondria by the action of a carrier that transports GSH from the cytosol to the mitochondrial matrix [42]. Upon exposure to increased exogenous ROS, isolated, perfused rat hearts are rapidly depleted of their antioxidant reserves, including those of SOD and GSH, rendering them more vulnerable to the action of oxidative injury [43].
Another important mechanism in the antioxidant protection is the sequestering of iron and copper ions to keep them from reacting with superoxide or H2O2. The antioxidant dexrazoxane prevents site-specific iron-based oxygen radical damage by chelating free and loosely bound iron. Hence, this compound has been used as a cardioprotective drug against doxorubicin-induced oxidative damage to myocardial mitochondria in both humans and animals [21, 44]. The antioxidant metal-binding protein metallothionein (MT) also provides cardioprotection by directly reacting with ROS produced by ischemia/reperfusion and doxorubicin treatment, as found in studies with a cardiac-specific MT-overexpressing transgenic mouse model [45]. MT expression is also inducible in the heart and other tissues by TNF-α, IL-6, doxorubicin, and metals such as cadmium and zinc [45–47]. However, whether MT plays cardioprotective role remains yet to be determined.
The uncoupling of mitochondrial respiration from ATP production, by either artificial uncouplers such as 2,4-dinitrophenol (e.g., DNP) or natural uncouplers (e.g., laurate), FA, and mitochondrial uncoupling proteins (UCP), strongly inhibits O2•− and H2O2 formation in mitochondria [48–50]. ROS production is favored when the mitochondrial membrane potential is above a specific threshold. Under conditions where the mitochondrial membrane potential is at its peak (e.g., state 4 respiration), ROS production is increased. It is noteworthy that increased mitochondrial membrane potential slows electron transport through the respiratory chain, resulting in increased half-life of the ubiquinone free radical and the likelihood that electrons will interact with oxygen to form ROS [51]. Uncouplers prevent the transmembrane electrochemical H+ potential difference (∆ym) from being above a threshold critical for ROS formation by respiratory complexes I and III. Inconsistent with this mechanism, transgenic mice deficient in UCP3 exhibit enhanced ROS production and increased OS in the heart and skeletal muscles [52], whereas transgenic mice with UCP1 overexpression [53] and cardiomyocytes with UCP2 overexpression display markedly attenuated ROS generation [54].
Pharmacological inhibition of xanthine oxidase-derived superoxide formation and neutralization of peroxynitrite or inhibition of poly(ADP-ribose) polymerase (PARP) have been reported to provide significant benefit in various forms of cardiovascular injury [55]. Using rat and mouse models of HF, the beneficial effects of a novel ultrapotent PARP inhibitor have been examined by Pacher et al. [56]. The authors have studied the effect of INO-1001 on the development of HF induced by permanent ligation of the left anterior descending coronary artery, HF induced by doxorubicin, and acute myocardial dysfunction induced by bacterial endotoxin. In the coronary ligation model, significantly depressed left ventricular performance and impaired vascular relaxation of aortic rings have been found; PARP inhibition has significantly improved both cardiac function and vascular relaxation. In the doxorubicin model, a single injection of doxorubicin has induced high mortality and a significantly decreased left ventricular systolic pressure, +dP/dt, −dP/dt, stroke volume, stroke work, ejection fraction, and cardiac output. Treatment with the PARP inhibitor has reduced doxorubicin-induced mortality and markedly improved cardiac function; on the other hand, PARP inhibition has not interfered with doxorubicin’s antitumor effect. In the endotoxin model of cardiac dysfunction, PARP inhibition attenuated the reduction in myocardial contractility elicited by endotoxin. Taken together, these data suggest that PARP inhibition may be a promising approach in the [experimental] treatment of various forms of acute and chronic HF.
HF patients have increased plasma catecholamine levels, and sympathetic stimulation may lead to increased production of ROS and possibly to endothelial damage/dysfunction and atheroma formation. Based on these observations, treatment with β-blockers and angiotensin-converting enzyme (ACE) inhibitors has been recommended. However, in HF, the potential beneficial antioxidative effects of carvedilol, a vasodilating β-blocker with antioxidant activity, are rather controversial. Whereas some groups have found a reduction of OS in HF patients treated with carvedilol [57], others have not [58]. Using immunohistochemistry, Nakamura et al. (150) [59] have assessed the expression of 4-hydroxy-2-nonenal (HNE)-modified protein, a major lipid peroxidation product, in endomyocardial biopsy tissues from 23 patients with DCM and 13 control subjects with normal cardiac function. They have also analyzed whether levels of lipid peroxides have been elevated in the myocardium of patients with dilated cardiomyopathy (DCM) and also if carvedilol reduces lipid peroxidation level. Significant upregulation of myocardial HNE-modified protein has been found in all myocardial tissue samples from patients with DCM compared to that of control subjects. In addition, biopsy samples from 11 patients with DCM have been examined before and after treatment with carvedilol. Following treatment, myocardial HNE-modified protein levels have decreased by 40%, and this decrease has been associated with improvement in HF. Taken together this study has confirmed that OS is elevated in HF and the administration of carvedilol results in reduction of OS and improvement in cardiac function. These investigators have also evaluated whether levels of 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage, are elevated in the serum and in myocardium of patients with DCM and whether carvedilol can decrease 8-OHdG levels [60]. DCM patients have shown significantly increased levels of 8-OHdG compared to control subjects. Following treatment with carvedilol, the serum levels of 8-OHdG in DCM patients have been decreased by 19%; this decrease correlates with improvement in HF. Thus, carvedilol seems to be an effective way to reduce the DNA oxidative damage.
Chin et al. [61] have assessed the antioxidative properties of carvedilol and ACE inhibitors in severe HF. The baseline indices for the measures of oxidative damage and endothelial function in 66 HF patients were significantly higher than healthy control subjects. After 3 months of maintenance therapy with β-blockers, a significant reduction in lipid hydroperoxides (LHP) levels occurred, but not in total antioxidant capacity (TAC), nor in plasma von Willebrand factor (vWf), an index of endothelial damage/dysfunction. ACE inhibitor therapy also significantly reduced vWf levels, but failed to have any statistically significant effects on LHP or TAC. This study suggests that OS in advanced, severe HF may be due to increased free-radical production or inefficient free-radical clearance by scavengers and that β-blockers, but not ACE inhibitors, decreased lipid peroxidation; no relation was found between a reduction in oxidative damage and endothelial damage/dysfunction.
Treatment with ACE inhibitors, angiotensin, aldosterone, and endothelin antagonists has been shown to beneficially modulate endothelial dysfunction in severe HF. As pointed out by Bauersachs and Widder [62], these therapies increase NO bioactivity by either modulation of ROS generation, thereby preventing the interaction of superoxide anions with NO and/or increasing endothelial NO synthase (eNOS) expression and/or activity. In rats after large myocardial infarction, treatment with AVE9488, a novel eNOS transcription enhancer, attenuates cardiac remodeling and endothelial dysfunction. Furthermore, antioxidants, L-arginine, cofactors of endothelial NO synthase, and exercise training positively modulate endothelial function [63].
Mitochondrial-Based Therapy of FAO Disorders, Dysrhythmias, and CHF
The treatment of disorders of mitochondrial long-chain FAO is based on avoidance of fasting and replacement of normal dietary fat by medium-chain triglyceride. Defects in CPT-II, carnitine acylcarnitine translocase, or mitochondrial trifunctional protein (MTP), part of mitochondrial FAO, can be treated with drugs targeted to enhance glucose and pyruvate use, at the expense of FAO, to prevent the accumulation of long-chain acylcarnitines that can result in cardiac conduction defects and dysrhythmias [3, 64]. In contrast, acute cardiomyopathy associated with very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, which can be diagnosed by acylcarnitine analysis even in the neonatal period, can be treated with dietary therapy including medium-chain triglycerides [65]. Long-chain FA accumulation and their side effects can also be effectively reversed by inhibition of CPT-I activity with perhexiline and amiodarone.
In the failing and ischemic heart, there is a plurality of changes in myocardial metabolism. Modulation of myocardial glucose and FA metabolism is recognized as a target for therapeutic intervention. The treatment of patients in CHF using carvedilol, a β-adrenoreceptor blocker, results in marked improvement in myocardial energy efficiency by shifting myocardial oxidative substrates from FA to glucose [66]. Free FA is a primary source of energy during cardiac ischemia and can also serve to uncouple OXPHOS and increase myocardial O2 consumption. On the other hand, inhibitors of FAO can increase glucose oxidation and may improve cardiac efficiency. It is noteworthy that inhibitors of FAO can help to prevent the hyperglycemia that occurs in noninsulin-dependent diabetes. Since the inhibition of FAO is effective in controlling abnormalities in diabetes, inhibition of FAO enzymes, such as CPT-I, may also prove useful in the treatment of diabetic cardiomyopathy. There are a number of FAO inhibitors including etomoxir, oxfenicine, perhexiline, aminocarnitine, trimetazidine, ranolazine, hypoglycin, and DCA [67, 68]. In animal models, etomoxir, an inhibitor of CPT-I, has reversed changes in fetal gene expression, preserved cardiac function, and prevented ventricular dilatation [69]. In clinical studies of HF patients, etomoxir has improved systolic ventricular function, increased ejection fraction, and decreased pulmonary capillary pressure [30, 70]. Ranolazine treatment reduces cellular acetyl-CoA content via partial inhibition of FAO (it is therefore termed a pFAO inhibitor) and activates PDH activity. Clinically, it has been used to treat both ischemia and angina [71]. This treatment increases ATP production, reduces the rise in lactic acidosis, and improves myocardial function under conditions of reduced myocardial oxygen delivery. Trimetazidine provides protective effects against myocardial ischemia, diabetic cardiomyopathy, and exercise-induced angina in several clinical and experimental investigations [67, 68, 72]. While initially, trimetazidine has been thought to be an inhibitor of the activity of the last enzyme involved in mitochondrial FAO, 3-ketoacyl coenzyme A thiolase [73], recent observations have cast doubt on FAO inhibition as the primary mechanism, by which trimetazidine mediates cardiac recovery [74]. Another related effect of trimetazidine, which may contribute to its anti-ischemic action, is the acceleration of phospholipid synthesis and turnover with significant consequences for α-adrenergic signaling [75].
Clinical studies have suggested that polyunsaturated FA (e.g., n-3 PUFA) or fish oil supplementation reduces mortality and sudden death associated with CHF [76]. Its effect on mortality and morbidity is being gauged in the GISSI Heart Failure project, a large-scale, randomized, double-blind study [77]. A somewhat smaller but carefully designed study has recently confirmed that n-3 PUFA treatment markedly reduces the incidence of both atrial and ventricular dysrhythmias [38] [78]. Among a large assortment of PUFA-mediated effects on cardiomyocyte membrane lipid organization and function, the incorporation of n-3 PUFA (normally associated with reduced arachidonic acid) induces reduction of mitochondrial FAO and oxygen consumption in the heart. These effects on mitochondrial metabolism are primarily shown during post-ischemic reperfusion as improved metabolic and ventricular function. Both aging and ischemia markedly decrease levels of n-3 PUFA and cardiolipin in myocardial membranes, effects which have been correlated with increased mitochondrial Ca2+ levels that can in turn affect various mitochondrial enzymatic activities [79].
Cardioprotective Agents
Animal studies have shown that specific drugs (e.g., protein kinase C inhibitors, adenosine receptor agonists [Table 23.2]) targeting different steps of the cardioprotection (CP) signaling pathways, applied at the immediate onset of reperfusion, can significantly reduce the size of myocardial infarct and improve cardiac function [80, 81]. It has also been shown that treatment with a particular pharmacological class of calcium antagonists (e.g., diltiazem and verapamil) can reduce a number of the harmful effects of calcium overload following myocardial ischemia and particularly prominent during early reperfusion, leading to recovery of myocardial contractility and restoration of the levels of critically needed myocardial high-energy phosphates [82, 83]. Clinical studies have also shown that diltiazem and verapamil treatments can be beneficial to patients after myocardial infarct and with cardiac dysrhythmias [84, 85]. Interestingly, the clinical benefits of reducing the size of myocardial infarct and increasing the viability and recovery of regional function appear to be significant if diltiazem treatment is applied prior to myocardial perfusion [86]. Diltiazem inhibits sodium-induced Ca2+ release in isolated mitochondria. The increased mitochondrial Ca2+ levels result in elevated Ca2+-induced dehydrogenase activities, increased respiration, and restored ATP levels [87]. Intravenous injection of diltiazem can be cardioprotective both as an anti-ischemic and anti-dysrythmogenic agent when infused in patients undergoing coronary artery bypass grafting [88].
Table 23.1
Metabolic and antioxidant treatment of mitochondrial-based cardiac disorders
Treatment | Primary mechanism | Disorder |
|---|---|---|
Coenzyme Q10 | Antioxidant/ETC carrier | Heart failure, FRDA, MELAS, KSS |
Dichloroacetate | Increase PDH activity Decrease FAO | KSS, MELAS, lactic acidosis |
Diabetic cardiomyopathy | ||
Idebenone | Antioxidant | Friedreich ataxia (FRDA), mitochondrial cardiomyopathy |
Carnitine | Increased fatty acid transport | Cardiomyopathy and heart failure |
Etomoxir | FAO inhibitor | FAO disorders |
Trimetazidine | FAO inhibitor, phospholipid turnover | FAO disorders |
Myocardial ischemia/angina, diabetic cardiomyopathy | ||
Ranolazine | Partial FAO inhibitor | FAO disorders |
Myocardial ischemia/angina | ||
Perhexiline | FAO inhibitor | Dysrhythmia |
n-3 PUFA | Reduced FAO | Dysrhythmia |
Copper supplement | Assist in COX subunit assembly | HCM due to SCO2 mutation |
Dexrazoxane | Antioxidant | Doxorubicin-induced cardiomyopathy |
Carvedilol | β-adrenergic blocker | Congestive heart failure |
FAO shift to glucose | ||
Diltiazem | Inhibits release of mitochondrial Ca2+ | Cardiac dysrhythmia |
Myocardial ischemia |
As noted previously, several clinical studies have shown that mitochondrial-based CP elicited by ischemic preconditioning (IPC), as well as by the use of physiological stress and pharmacological stimuli (e.g., exercise, adenosine), can provide beneficial results in treating angina [89, 90]. Moreover, in recent clinical trials, the chronic administration of the mitoKATP opener nicorandil has been shown to improve the prognosis of patients with coronary artery disease [91]. The further use of CP-based strategies in treating patients undergoing angioplasty in ischemic cardiomyopathy, heart transplant, and bypass surgery is currently under consideration.
Volatile anesthetic agents commonly used to maintain the state of general anesthesia, such as halothane, isoflurane, and sevoflurane, can provide CP in response to myocardial ischemia and reperfusion [92–94]. The cardioprotective signaling pathway of anesthetic preconditioning (APC) shares components with IPC including protein kinase C activation, mitoKATP channel activation, and mitochondrial ROS generation. However, some differences between APC and IPC exist. The direct inhibition of mitochondrial ETC enzymes and altered mitochondrial bioenergetics in hearts preconditioned by APC implicate the mitochondria as a primary target. Decreased mitochondrial ROS levels in ischemic and reperfused hearts preconditioned by APC have been proposed to contribute to facilitate the maintenance of mitochondrial structure and function [94]. Clinical studies have confirmed that sevoflurane preconditioning, assessed by biochemical markers, preserves myocardial function in patients undergoing coronary artery bypass graft surgery under cardioplegic arrest [95].
Another potential target of CP is the apoptotic pathway, resulting from the permeabilization of mitochondrial membranes, leading to the release of proapoptotic proteins and to bioenergetic failure. Mitochondrial apoptosis, which can also result from myocardial ischemia/reperfusion, plays a pivotal role in the progression of myocardial remodeling in hypertrophic cardiomyopathy (HCM) and DCM. Attenuation of the mitochondrial apoptotic pathway in cultured cardiomyocytes by overexpression of the anti-apoptotic protein Bcl-2 or in animal models by treatment with antioxidants (e.g., melatonin) has been shown to provide CP [96, 97]. Modulation of MPTP opening, a common early event in the mitochondrial apoptotic pathway, can be directly mediated by cyclosporin A or sanglifehrin A treatment providing CP against reperfusion injury [98]. Uncouplers of OXPHOS such as dinitrophenol and CCCP have also been shown to elicit CP [99, 100]. Furthermore, overexpression of the UCP2 in cultured neonatal rat cardiomyocytes has suppressed markers of apoptotic cell death, prevented the loss of mitochondrial membrane potential, and attenuated both mitochondrial Ca2+ overload and ROS production, protecting cardiomyocytes exposed to OS [54]. Thus, the discovery that potassium channel openers (KCOs) diazoxide and pinacidil facilitate proton translocation through mitochondrial membranes acting as uncouplers of OXPHOS, activating state 4 respiration and depolarizing the mitochondria, is not surprising [101]. However, the majority of cardioprotective treatments targeting myocardial apoptosis (e.g., modulation of uncoupler or MPTP activity) can have significant impact on a variety of metabolic processes; therefore, their therapeutic application may prove to be problematic. Notwithstanding, in preliminary clinical studies several reagents (mostly antioxidants) have shown promising results. In a group of patients undergoing cardiac surgery with cardioplegic arrest, increased myocardial apoptotic progression has effectively been prevented in those cases infused with N-acetylcysteine, an antioxidant and sulfhydryl donor precursor for glutathione [102]. In addition, grape seed proanthocyanin extract (GSPE), a potent antioxidant, showed cardioprotective properties in both animal and human, improving post-ischemic left ventricular function, significantly reducing infarct size, myocardial ROS levels, and apoptotic markers [103]. This finding confirms previous observations, which have documented that the polyphenolic antioxidants present in red wine, such as resveratrol and proanthocyanidins, provide CP by their ability to function as in vivo antioxidants in addition to the alcoholic component, which also is cardioprotective by adapting the heart to OS [65–66] [104, 105]. Pyruvate also provides CP both in animal models [98, 106] and in patients [107], although the precise mechanism has not yet been determined.
A complementary approach to cardioprotective therapies targeting apoptosis involves triggering anti-apoptotic cell proliferation (or cell-survival pathways). Various growth factors, including insulin-like growth factor (IGF-1), hepatocyte growth factor, endothelin-1, fibroblast growth factor, and transforming growth factor, have been shown to protect the heart against OS, largely by attenuating cardiac myocyte apoptosis [108]. Growth factor signaling, such as induced by IGF-1, is mediated via activation of the PI3K pathway, which has been shown to be cardioprotective, and by the serine-threonine kinase, Akt. However, concerns have been raised by the systemic administration of high levels of IGF-1 peptide. In vivo cardiac IGF-1 gene transfer, performed prior to ischemia/reperfusion injury, has led to a more sustained activation of Akt and reduced hypoxia-induced apoptosis compared to IGF-1 peptide treatment [109]. Somatic gene transfer of growth factors may be advantageous over systemic delivery by mediating cardiomyocyte protection without elevating serum levels of growth factors. Studies in transgenic animals with Akt overexpression have shown a variety of cardiac phenotypes including progressive cardiac hypertrophy and failure. Thus, considerable caution is warranted in the therapeutic application of modulation of Akt signaling since it is a critical mediator of hypertrophic growth [110, 111]. However, in transgenic animals, hypertrophic remodeling resulting from Akt myocardial overexpression can be eliminated by nuclear targeting of Akt, which enhances survival of cardiomyocytes with no loss of CP vs. ischemia [73] [112].
Similarly, a “metabolic cocktail” composed of glucose-insulin-potassium, when administrated at early reperfusion, reduces infarct size in the rat heart in vivo, representing a relatively inexpensive approach to CP, in which the insulin component primarily stimulates Akt prosurvival signaling [113]. Since intravenous insulin therapy is associated with metabolic side effects, the development of therapeutic agents that can target downstream cell-survival insulin-activated signaling events has been considered as an alternate approach to promote CP [114].
Animal Models of Mitochondrial-Based Heart Disease
One of the most informative and successful tools to identify the molecular cause of mitochondrial disorders has been transgenic animals. (A general overview of cellular and animal models and the growing list of transgenic mouse models has been presented in Chap. 24 [Table 23.3].) The development of animal models for mitochondrial-based cardiac disease has been extremely useful in highlighting the multiple pathways that (if perturbed) can lead to dysfunction in the human heart. It is also invaluable for the identification of potential targets for future drug and gene therapies as well as for the evaluation of the efficacy of novel treatment strategies [115, 116]. Null mutations in a number of nuclear-encoded genes involved in mitochondrial metabolic function can lead to cardiac dysfunction in transgenic mice (Table 23.3). Unfortunately, null mutations which are easily generated in the mouse have not yet been generated in other animal species (e.g., rat, dog, pig). However, strategy based on the overexpression of specific genes has been exploited in a variety of organisms and has proved to be extremely informative in delineating mechanisms mediating the regulation of cardiac function and onset of cardiac disease.
Table 23.2
Cardioprotective agents
Agent | Mechanism |
|---|---|
Ischemic preconditioning | Activates CP pathway |
Nicorandil | MitoKATP channel opener |
Sevoflurane | Volatile anesthetic |
N-acetylcysteine | Antioxidant; glutathione precursor |
Pyruvate | Not determined |
Proanthocyanin extract | Antioxidant |
Phosphorylated GSK-3β | Inhibits MPTP opening |
Glucose-insulin-potassium | Akt activation |
Adenosine | Activates CP pathway |
Moderate alcohol | Akt activation |
Other polyphenols | Antioxidant |
Diazoxide, pinacidil | MitoKATP channel opener |
Bcl-2 overexpression | Apoptotic inhibitor |
Cyclosporin A | MPTP modulator |
Dinitrophenol, CCCP | Uncouplers |
Table 23.3
Transgenic animal models of mitochondrial-based disease
Gene | Cardiac phenotype | References |
|---|---|---|
Nuclear gene knockouts | ||
Adenine nucleotide translocator (ANT1) | Cardiomyopathy and defective coupled respiration | [117] |
Mn superoxide dismutase (SOD2) | Cardiomyopathy and SOD deficiency | [118] |
Heart-specific mtTFA | DCM and cardiac conduction defects, mtDNA depletion, and respiratory chain defects | [119] |
Frataxin | Cardiomyopathy and cardiac hypertrophy; respiratory chain defect; Fe-S deposits | [120] |
Long-chain acyl-CoA dehydrogenase (LCAD) | Cardiomyopathy and sudden death; impaired FAO | [121] |
Mitochondrial trifunctional protein alpha subunit (MTPα) | Cardiac necrosis and sudden death; lipid accumulation with impaired FAO | [122] |
Very long-chain acyl-CoA dehydrogenase (VLCAD) | Increased ventricular tachycardia, lipid and mitochondrial accumulation | [123] |
RXR α | Embryonic HF with defects in ETC, ATP levels, and increased mitochondria | [124] |
MEF2A | DCM and sudden death with severe mitochondrial disorganization and dysfunction | [125] |
Mitochondrial DNA | ||
Chloramphenicol resistance (CAP R) | Cardiomyopathy and perinatal death | [126] |
4.7 kb deletion | Cardiomyopathy and respiratory chain defect | [127] |
Mitochondrial proteins expressed by engineered genes may allow the correction of specific enzymatic/genetic mitochondrial defects [128, 129]. However, there are limitations in the repertoire of mitochondrial genes which can be functionally replaced in mammalian cells by allotopic expression [130].
Given the systemic nature of many mtDNA defects, a major difficulty is how to deliver a functional agent (e.g., gene or gene product) into mitochondria for repair of impaired function [131]. Strategies to address this problem include the transplant of healthy mitochondria into the germ line. This can be achieved by transfer of a nucleus from a fertilized oocyte to a healthy donor cytoplast. Several techniques have been used to successfully introduce genetically distinct mtDNA molecules into the mouse female germ line. Heteroplasmic mice have been created by the fusion of cytoplasts generated from mouse ova with single zygotes [132] by fusion of a zygote nucleus and a portion of the oocyte cytoplasm with enucleated eggs [133]. Moreover, it could be done by the microinjection of somatic cell mitochondria from one species of mice into zygotes of another species generating xenocybrids [134]. The fusion of cytoplasts heteroplasmic for a 4,696-bp mtDNA deletion to pronucleus-stage zygotes has also been used to generate the first mouse model of mtDNA disease [127], including the development of cardiomyopathy. The fusion of cytoplasts to undifferentiated mouse female embryonic stem (ES) cells has also been used to introduce a well-characterized mouse mtDNA mutation, the 16S rRNA mutation resulting in chloramphenicol resistance (CAPR) into the mouse female germ line [126, 135]. Mice homoplasmic for the CAPR mutation exhibited myopathy, DCM, and perinatal or in utero lethality, validating the ES cell approach to produce transmitochondrial mice. More recently, the strategy of xenocybrid transfer of mitochondria by cytoplast fusion, combined with the use of mtDNA-depleted ES cells, enabled the generation of transmitochondrial mice with germline transmission of homoplasmic mitochondria containing the introduced alleles [136].
The use of transmitochondrial oocytes in human studies has a limited and controversial history. Ooplasmic transplantation has been used in several studies in conjunction with in vitro fertilization clinics [137, 138]. In these reports, the addition of a small amount of injected ooplasm, derived from fertile donor oocytes, into developmentally compromised oocytes from patients with recurrent preimplantation failure has been demonstrated to enhance embryo viability and led to the birth of 15 children. The mtDNA from the donor as well as the recipient cell mtDNA has been found to be present in blood of the child emerging from the transplanted oocyte at 1 year of age. Excluding the numerous ethical considerations provoked by this first case of human germline genetic modification, several cautions have been called for by these studies including the potential for long-term harm in chromosomal segregation and aberrant division, predicted by similar studies conducted in lower organisms, and epigenetic influences of foreign cytoplasm demonstrated in numerous studies of cytoplasmic transfer in mice [139]. In fact, 2 of the 15 pregnancies have resulted in unexpected chromosomal abnormalities, including Turner syndrome. The long-term deleterious influence of heteroplasmic mtDNA has also been considered as a potential problem in this approach [140]. If we are to fully appreciate the outcomes associated with embryo manipulation, then extensive investigations with animal models that incorporate genetic, biochemical, and physiological analyses are necessary, accompanied by clinical monitoring, to demonstrate the suitability of these techniques for human use.
In patients with mitochondrial disease due to specific mtDNA defects, recommendations have been formulated regarding the use of prenatal diagnosis, including preimplantation diagnosis and chorionic villus sampling [141]. Prenatal diagnosis of defects such as Mendelian defects and syndromes caused by mutations at nt 8993 appears to be more reliable than with other maternally inherited defects, in which there is little correlation found between phenotype and the amount of mutant allele (mutant load).
Mitochondrial Defects and Gene Therapy
Identification of Genetic Defects
Breakthroughs in molecular genetic technology stemming from the use of chromosomal mapping and identification of genes involved in both the primary etiology and also as significant risk factors in the development of cardiac and vascular abnormalities have been used. Specific genetic and molecular factors linked to congenital heart defects (CHD) and cardiac dysrhythmias may allow their identification providing a unique opportunity to improve genetic diagnostics and the potential use of gene therapy to treat cardiovascular disease (CVD) (Fig. 23.1).
Many of the nuclear gene defects implicated in cardiomyopathies were originally mapped by linkage analyses in affected families, allowing the subsequent identification of candidate genes (and mutant alleles) by positional cloning and nucleotide sequence analysis. Molecular techniques including polymerase chain reaction (PCR), restriction fragment length polymorphism (RFLP), and single-strand conformation polymorphism (SSCP) have been used in screening defective alleles from the proband and family members to establish inheritance patterns. In many cases, detection of novel nuclear mutations by itself is a very difficult undertaking involving a thorough analysis of multiple, large coding regions (exons) of one, if not more, candidate gene. This is significantly less problematic with the shorter, intron-less mtDNA genes. Moreover, in the relatively well-characterized cases of familial HCM gene screening, the consensus has been reached that each specific HCM-causing mutation is rare, challenging the view of common mutations since most families have “private” or novel mutations [142]. While somewhat true of mtDNA mutations, molecular “hotspots” (e.g., tRNALeu and cytb genes) appear to be more likely affected in mitochondrial cardiomyopathies [143]. Correlation of the clinical course and prognosis with specific mutations may be informative with nuclear gene mutations; for instance, specific β-MHC mutations in HCM are associated with a high incidence of sudden death, whereas other mutations are not. On the other hand, specific mtDNA gene mutations are more frequently associated with variable clinical phenotypes, and modifying nuclear factors influencing the expression of pathogenic alleles have often been proposed.
Detection of pathogenic mutations has highly been facilitated by the development of high-throughput analytical techniques like denaturing high-performance liquid chromatography (DHPLC) or capillary array electrophoresis, which will further advance the use of molecular genetic analysis in clinical and preclinical diagnosis [144]. Moreover, the availability of gene chip technology may allow in the near future not only the automated and rapid screening of mtDNA and nuclear gene mutations from clinical samples, but also the assessment of their impact on specific myocardial gene expression. The presence of multiple modifier genes significantly influences the phenotypic expression and severity of pathogenic HCM genes [145]. Maternally inherited disorders with variable penetrance, often restricted to tissue-specific expression (i.e., LHON [mitochondrial non-syndromic sensorineural hearing loss] and a form of mitochondrial HCM), have been proposed to be caused by a primary homoplasmic mtDNA pathogenic mutation acting in concert with a nuclear modifier, which can be a common functional polymorphism in tissue-specific protein, possibly with mitochondrial location [146]. In this disease model, the mtDNA mutation, although necessary, is not sufficient to induce the pathology, whereas the nuclear modifier does not induce any pathology per se, but contributes to the pathogenic effect of the mitochondrial mutation. Identification of modifier genes, which will markedly improve the detection of genetic risk factors, has been facilitated by large-scale genome-wide approaches in an effort to identify polymorphic variants correlated with disease severity.
Gene Therapy
Identification of genes affected in CVD has lead to the development of improved therapies, based on the use of gene replacement and/or gene suppression (silencing). Preclinical studies in several animal models have shown that gene therapy can be beneficial in the treatment of HF, hypertension, hypertrophy, cardiac dysrhythmias, and myocarditis as well as in disorders of the vascular wall, particularly in cases where drug therapy has proved to be of limited value. Gene therapy enables therapeutic concentrations of a gene product to be accumulated and maintained at optimal levels and at a localized target site of action. It also offers the possibility of minimizing systemic side effects by avoiding high plasma levels of the gene product [147]. While early phases of clinical gene therapy trials for CVD have shown promising results, mainly in regard to therapeutic angiogenesis and restenosis treatment, the development of improved vectors and safe and efficient delivery methods and the acquisition of safety and toxicity data need to be improved before these therapies can be routinely used in a clinical setting.
Both viral and naked plasmid DNA vectors have been employed in preclinical and clinical cardiovascular gene transfer studies. While plasmid DNA vectors have been shown to have good entry and expression in normal and ischemic muscle [148], their lower efficiency of transfection in myocardial gene delivery has limited their use. Features of viral vectors can predetermine both the range of host cells that can be transduced and the efficiency, level, and duration of transgene expression. Adenoviral vectors can transduce both dividing and nondividing cells and are particularly efficient in transfecting postmitotic cells including cardiomyocytes and, to a lesser extent, vascular cells and therefore have been the primary viral vector of choice. A limitation of the adenoviral vectors is their provision of transient rather than prolonged transgene expression. Moreover, adenoviral vectors pose additional safety concerns; these vectors produce increased inflammation, and long-term cell- and antibody-mediated immune responses have been widely reported [149]. Nevertheless, to date, no evidence of serious adverse effects has been reported in clinical trials of cardiovascular gene therapy using adenoviral vectors involving over 150 subjects [150]. Other viral vectors are being considered for the use in cardiovascular therapies including lentivirus and recombinant adeno-associated virus (AAV). AAV is taken up more slowly into myocardial cells. Compared to adenovirus, the levels of AAV transgene expression are lower but can be longer in duration, being sustained in rodent myocardium for 9–12 months. Moreover, AAV vectors have a lower potential to induce unwanted inflammation or immunocytotoxicity [147].
Another alternative gene transfer approach involves the use of antisense strategies. The use of antisense oligonucleotides or small interfering RNA (siRNA) allows to regulate the transcription of targeted endogenous genes by selectively inhibiting their expression.
The antisense oligonucleotide approach can employ either single-strand or double-strand oligonucleotides to target specific gene expression, whereas siRNA involves the use of a specific double-strand RNA construct to silence specific gene expression (RNAi). Double-strand oligonucleotides homologous to the cis-regulatory sequences of the promoter of a gene of interest can be similarly employed. They can function as molecular decoys to bind specific transcription factors and therefore block the expression of genes encoding those transcription factors [151]. A similar strategy has been employed to block cell cycle progression to modulate cell proliferation.
In addition to several well-characterized animal models of cardiac gene therapy aimed at treating restenosis, hypertension, and angiogenesis [152], a number of nuclear gene targets to elicit increased myocardial protection and improve cardiac function have been described.
Short-term protection of the heart from ischemia and OS can be provided by gene transfer and overexpression of genes encoding critical antioxidant enzymes such as SOD or heme oxygenase (HO-1). Introduction of a myocardial protective gene, such as HO-1, employing a recombinant AAV vector, into myocardium prior to coronary artery ligation, has significantly reduced infarct size in a rat model of ischemia and reperfusion [153]. In addition, gene-mediated CP against myocardial ischemia has been achieved by introducing and overexpressing genes for the free-radical scavenger enzyme SOD [154], molecular chaperone HSP70 [155], and anti-apoptotic mitochondrial protein Bcl-2 [156]. It remains to be seen whether this strategy can provide long-term CP against repeated, chronic forms of ischemic insult.
Experimental cardiac gene therapy has also provided useful information in cardiomyopathy and HF. Transgenic mice lacking desmin, the muscle-specific member of the intermediate filament gene family, develop cardiomyopathy characterized by extensive cardiomyocyte death, fibrosis, and eventually HF. There is evidence that mitochondrial abnormalities are implicated in the onset of the cardiomyopathy. The overexpression of the Bcl-2 in the desmin null heart resulted in the correction of mitochondrial defects, reduction in the occurrence of myocardial fibrotic lesions, prevention of cardiac hypertrophy, restoration of cardiomyocyte ultrastructure, and significant improvement of cardiac function [157].
Nuclear-encoded gene products affecting mitochondrial metabolism have also recently proved to be an effective target for gene therapy in the rat central nervous system (CNS) and human fibroblasts. The E1α subunit of the pyruvate dehydrogenase complex (PDHC) has been successfully transduced using an AAV construct; transduction of cultured fibroblasts from a patient with an E1α deficiency led to a partial restoration of PDH activity [158]. Given the pivotal role of PDHC in the regulation of aerobic metabolism, the delivery and/or modulation of this gene in cardiac tissues may prove to be useful in treating disorders, in which cardiac aerobic metabolism is affected including ischemia, hypertrophy, and HF.
Targeting the Mitochondria Using Nucleic Acids
Gene therapy to replace or repair defective mitochondrial genes could be an important adjunct in the treatment of mitochondrial-based CVD. However, it has not yet been proven possible to introduce and replace (or repair) mtDNA genes in the mitochondria of either in vitro cultured cells or more importantly in the organelles of in vivo myocardium, posing a major hurdle for gene therapy of mtDNA-based disorders. While biolistic transformation using highly accelerated DNA-coated metal particles has successfully been employed to deliver genes into bacteria, and into the organelles of plants and yeast, this technique has not been proven to be applicable in the transformation of mammalian mitochondria. Another approach, electroporation of nucleic acids, while effective in the delivery of genes to the nucleus, with their subsequent expression, has not been successfully applied to the gene delivery and expression in mitochondria of living cells [159]. In addition to the difficulties associated with a delivery system for mitochondrial genes, the replacement of endogenous multiple-copy defective genes (within multiple organelles) also poses a significant challenge. Nonetheless, despite the present lack of a reliable mitochondrial transformation system, several approaches applied primarily to isolated cells of individuals affected with mitochondrial diseases or from cybrids have shown promising results [Marín-García et al., unpublished data] [131, 160] Table 23.4. These include the selective destruction of mutant mtDNA by importing a restriction endonuclease enzyme into mitochondria [161], replacement of a mutant mtDNA-encoded protein with a genetically engineered wild-type equivalent encoded and expressed by nuclear genome (also called allotopic expression) [128, 129], or using gene replacement of defective mtDNA alleles with cognate genes transfected from other organisms [162].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


