Chapter 41
Systemic Complications
Renal
Daniel J. Myers, Stuart I. Myers
One must have a basic comprehension of renal anatomy and physiology to begin to understand the complex changes in renal function that can occur during treatment of vascular disorders. This chapter can provide only a brief summary of renal function and physiology, but more detail can easily be obtained from several standard texts.1
Renal Anatomy
The specific components of the kidney include the nephrons, the collecting ducts, and the microvasculature. The human kidney contains approximately 1 million nephrons, which are established before birth. Injured or lost nephrons cannot be replaced, so preservation of nephrons is essential to maintain normal renal function. There are two basic types of nephrons, those with short loops of Henle and those with long loops. The long loops extend to the inner medulla and the short loops to either the outer medulla or the cortex. The collecting ducts descend within the medullary rays of the cortex, and on entering the inner medulla, fuse to form papillary ducts (Fig. 41-1A).
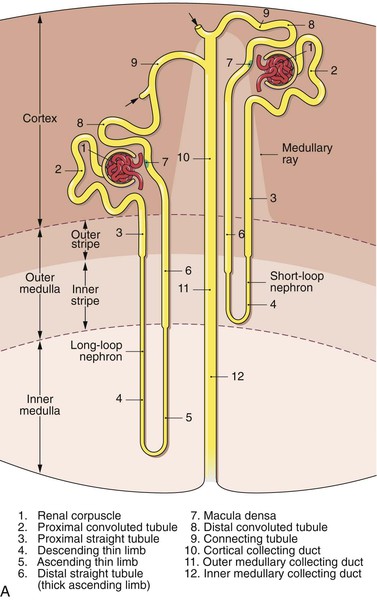
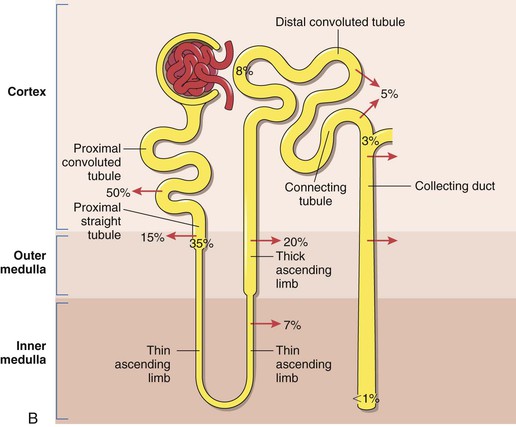
Figure 41-1 A, Nephrons and the collecting duct system. Short- and long-looped nephrons are shown together with a collecting duct (not drawn to scale). Arrows denote the confluence of further nephrons. (Adapted from Kriz W, et al: Renal anatomy. In Floege J, Johnson R, Freehally J, editors: Comprehensive clinical nephrology, ed 4, Philadelphia, PA, 2010, Mosby Elsevier, p 4.) B, Renal sodium handling by the nephron. Figures outside the nephron represent the approximate percentage of the filtered load reabsorbed in each region. Figures within the nephron represent the percentages remaining. (Adapted from Shirley DG, et al: Renal physiology. In Floege J, Johnson R, Freehally J, editors: Comprehensive clinical nephrology, ed 4, Philadelphia, PA, 2010, Mosby Elsevier, p 21.)
The renal artery divides into the interlobar arteries, which enter the cortex. The interlobar arteries divide into the arcuate arteries at the junction of the cortex and the medulla. These vessels then divide into the cortical radial arteries, which ascend through the cortex. No arteries penetrate the medulla. The cortical radial arteries give rise to the afferent arterioles, which supply the glomeruli. The glomeruli are drained by efferent arterioles that can be cortical or juxtamedullary. The cortical efferent arterioles are derived from the superficial and midcortical glomeruli and supply the capillary plexus of the cortex. The juxtamedullary efferent arterioles supply the renal medulla. At the level of the outer stripe of the medulla, the juxtamedullary efferent arterioles divide into the descending vasa recta, and from these individual vessels, leave at different levels to supply the adjacent medullary plexus. Ascending vasa recta drain the renal medulla and ascend as unbranched vessels. They then ascend and traverse the outer medulla and form the capillary plexus of the outer stripe. The ascending vasa recta then empty into the arcuate veins.
The microvasculature provides a unique countercurrent exchange between the blood entering and the blood leaving the medulla. The unique vascular anatomy also separates blood flow in the inner stripe from blood flow in the inner medulla. The descending vasa recta supplying the inner medulla are not exposed to the tubules of the inner or outer stripe (outer medulla). Blood flow in the ascending vasa recta from the inner medulla and inner stripe perfuses the outer stripe. The physiologic significance of this anatomy has been postulated to be a mechanism to prevent loss of solute from the medulla. Venous drainage accompanies the arteries. The arcuate veins form anastomosing arches at the corticomedullary border and are fed by veins from the cortex and medulla. The arcuate veins join to form the interlobar veins, which run parallel to the interlobar arteries (see Fig. 41-1A).2
Renal Function
The kidney is the major site responsible for maintenance of intravascular volume and composition under normal conditions. Toward this end, the kidney provides three basic physiologic processes: glomerular filtration, selective tubular secretion, and selective tubular reabsorption.3,4 The kidney receives a fourth of the cardiac output, and based on a cardiac output of 5 L/min, the kidney receives approximately 900 L/day of plasma flow. The glomeruli filter 20% of renal plasma flow and must reabsorb 99% of the 180 L of plasma filtered per day to provide a urine output of 1.8 L/day (for a 70-kg man). This ultrafiltrate has the same electrolyte and solute concentration as plasma, all of which is almost totally reabsorbed.5 The solute content and volume of urine that enters the renal pelvis are very different from the solute content and volume of the glomerular filtrate. The filtrate flows through the different portions of the tubule, where the solute content and volume are altered by tubular reabsorption and tubular secretion. When the direction of solute movement is from the tubular lumen to the peritubular capillary plasma, the process is termed tubular reabsorption. Movement in the opposite direction is termed tubular secretion. For any particular substance, there is a unique combination of filtration, reabsorption, and secretion (although not all substances are handled by all three processes). This is a dynamic process, in that the combination of filtration, secretion, and reabsorption is under physiologic control, so the relative rates of each process can be altered. For example, if one drinks a large quantity of water, the excess water will be excreted within 1 to 2 hours because of an increase in the glomerular filtration rate (GFR) and a decrease in the tubular reabsorption of water.
The descending loop of Henle is permeable to water but only minimally permeable to sodium and chloride, whereas the ascending loop is not permeable to water but has active transport mechanisms that readily transport the chloride ion with concomitant passive transport of sodium. This is the underlying basis for the countercurrent mechanisms that produce the medullary osmotic gradient that is important in the regulation of urine osmolarity. The sodium ion is also actively reabsorbed in the distal tubule. Reabsorption of sodium from the distal tubule and from the proximal collecting ducts is controlled by aldosterone secretion. The mechanisms responsible for sodium reabsorption by the kidney are remarkable, in that only 50 to 200 mEq of sodium is excreted per day of the 25,000 mEq of sodium filtered daily by the kidney (Fig. 41-1B). Potassium is reabsorbed in the proximal convoluted tubule and the thick ascending limb of Henle. Ten percent of the filtered load reaches the early distal tubule. Potassium secretion by connecting cells in the late distal tubule and/or cortical collecting system is variable and represents the major determinant of potassium excretion.3–6
Neuroendocrine Modulators of Renal Function
Intravascular volume is regulated primarily by a series of stretch receptors or baroreceptors located in the arterial tree and the atria. Because these receptors not only sense changes in pressure or volume (atrial receptors) but also monitor the rates of change during the cardiac cycle, they govern the effective circulating volume. Factors that decrease cardiac performance alter intravascular volume and are perceived by these receptors, which then alter renal function to retain salt and water, and thereby, increase the effective circulating volume. Similarly, when the concentration of circulating plasma proteins is reduced, there is a net diffusion of intravascular water into the extravascular space secondary to the decreased intravascular oncotic pressure. This net decrease in circulating volume is sensed by these same receptors, and neuroendocrine regulators of urinary output inhibit excretion of water to correct the volume deficiency. When the baroreceptors perceive a reduction in circulating volume, their afferent signals are reduced, which decreases their tonic inhibition over the neuroendocrine system. This reduced inhibition leads to increased secretion of vasopressin, 13-endorphins growth hormone, adrenocorticotropic hormone through the central nervous system, and an increased release of epinephrine from the adrenal medulla.
At the local renal level, a reduction in arterial pressure or central venous pressure (or both) results in an increase in renal sympathetic nerve stimulation that reduces urinary sodium excretion via three mechanisms: (1) constriction of afferent and efferent arterioles, which reduces renal blood flow and GFR; (2) reabsorption of sodium in the proximal tubule and the thick ascending loop of Henle; and (3) stimulation of renin secretion. At the level of the nephron, baroreceptors within the macula densa cells of the juxtaglomerular apparatus perceive a decrease in intravascular pressure or plasma ion concentration and stimulate juxtaglomerular cells to release renin. Renin is an enzyme that is stored in the specialized macula densa cells that constitute part of the juxtaglomerular apparatus. It is released in response to an increase in sympathetic nerve activity, reduced stretch of the afferent arteriole, and decreased transport of NaCl to the macula densa. Renin catalyzes the release of angiotensin I from angiotensinogen. Angiotensin I is then converted to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II has several important actions. First, it induces arteriolar constriction of the afferent and efferent arterioles and thereby causes an increase in blood pressure while decreasing renal blood flow; second, it stimulates renal sodium reabsorption in the proximal tubule; and third, it induces secretion of aldosterone from the zona glomerulosa of the adrenal cortex. Aldosterone secretion results in sodium reabsorption in the distal tubule and collecting duct.1–6
Paracrine and Endocrine Modulators of Renal Function
A number of endogenous and exogenous paracrine and endocrine substances influence renal function. An exhaustive list of these substances is beyond the scope of this chapter, but the major substances thought to be involved in normal and abnormal renal physiology are briefly presented. Eicosanoids are an important class of vasoactive metabolites of three different enzymes: cyclooxygenase (COX), lipoxygenase, and cytochrome P-450. COX is present as both constitutive (COX-1) and inducible forms (COX-2). Conversion of arachidonic acid to prostaglandins and thromboxane occurs via both enzymes. The major renal eicosanoids are prostaglandin E2 (PGE2) and prostaglandin I2 (PGI2). These potent vasodilators buffer renal vasoconstrictors such as thromboxane A2, angiotensin II, and norepinephrine. Decreased synthesis of the renal vasodilators PGE2 and PGI2 has been associated with renal vasoconstriction and has been demonstrated after a number of injuries, including renal ischemia, renal ischemia-reperfusion, and intestinal ischemia-reperfusion.7–15 PGE2 has been shown to inhibit sodium reabsorption from the thick ascending limb of Henle, and thus, contributes to protecting the renal medulla during hypoxia. COX-2 expression has been found in the macula densa and is thought to contribute to release of renin via PGE2. This could be one of the mechanisms responsible for the low renin levels found in patients taking nonsteroidal anti-inflammatory drugs (NSAIDs). The loss of COX-2 expression can also reduce medullary blood flow and cause apoptosis of medullary interstitial cells.
Nitric oxide (NO) is another potent endogenous renal vasodilator that has been shown to contribute to the maintenance of normal renal blood flow and function under normal physiologic conditions.12–23 Zou and Cowley24 demonstrated that NO was synthesized in both the medulla and the cortex.23 They concluded that NO might play a role in the control of vascular tone and tubular function in the kidney.23 Loss of endogenous NO synthesis has been suggested to contribute to renal vasoconstriction after renal ischemia-reperfusion injury from either local or systemic causes.12,13,16–24 Similar to the vasodilator prostaglandins, NO is present in the macula densa as the neuronal synthase form (nNOS). Downregulation of nNOS may contribute to arteriolar vasoconstriction by downregulating COX-2 (and subsequent PGE2 synthesis) through an adenosine or adenosine triphosphate (ATP) pathway. NO has also been shown to help mediate the increased natriuresis following an increase in renal interstitial hydrostatic pressure (caused by acute increases in blood pressure) without incurring a transforming growth factor (TGF)-mediated decrease in GFR.25 Atrial natriuretic peptide is released from atrial myocytes after atrial stretch secondary to increased blood volume. Atrial natriuretic peptide increases sodium excretion directly and by downregulating renin and aldosterone secretion.
Endothelins are a potent family of vasoconstrictor peptides. The kidney is a rich source of endothelins, which function as autocrine and paracrine substances. Endothelin-1 (ET-1) is synthesized in the afferent and efferent arterioles, where it induces vasoconstriction, and in mesangial cells, where it induces contraction. Upregulation of ET-1 synthesis induces a marked reduction in renal blood flow and GFR. ET-1 can inhibit sodium reabsorption in the medullary thick ascending limb, and this may, in part, be mediated by NO. The remaining role of endothelins in renal physiology is unclear at this time.
Purines are another complex class of autocrine substances that may be involved in renal physiology. Purinoreceptors are divided into P1 and P2 receptors. P1 receptors are upregulated in response to adenosine. P2 receptors are stimulated by nucleotides such as ATP and adenosine diphosphate (ADP). P2 receptors are subdivided into P2X (ligand-gated ion channels) and P2Y (metabotropic) receptors. P1 and P2X receptors are located in the afferent arteriole and contribute to vasoconstriction. P1, P2X, and P2Y receptors are located along the nephron. Endogenous adenosine enhances proximal tubular reabsorption, whereas luminal ADP inhibits it. In the collecting duct, basolateral ATP inhibits vasopressin-sensitive water reabsorption, and basolateral and luminal ATP inhibits sodium reabsorption. There is also evidence that the uric acid (end product of purine metabolism), may cause renal vasoconstriction, possibly by inhibiting release of NO and stimulation of renin.26 Further studies will provide insight into the role of purines in renal physiology.2
Renal Dysfunction After Vascular and Endovascular Procedures
Renal dysfunction after vascular and endovascular procedures can vary from mild natriuresis to fulminant acute tubular necrosis (ATN) and acute renal failure (ARF), requiring dialysis. Postoperative renal dysfunction can be classified as prerenal, renal, or postrenal (Fig. 41-2). Although the incidence of renal dysfunction complicating vascular and endovascular procedures has decreased with the development of appropriate perioperative fluid resuscitation, better surgical and endovascular techniques, and less nephrotoxic radiocontrast agents, the mortality associated with ARF remains high, ranging from 10% to 80% depending on the associated presence of multiorgan system failure.27,28
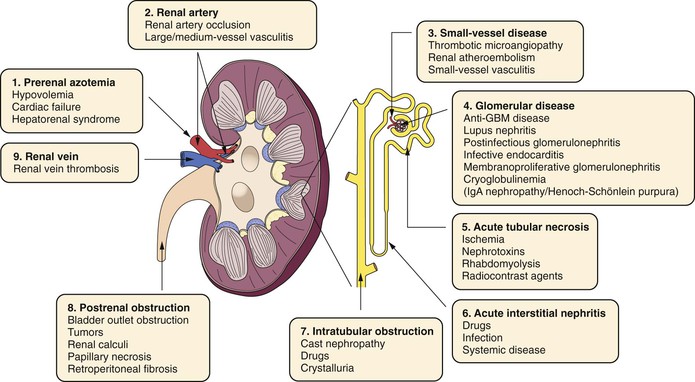
Figure 41-2 Causes of acute renal failure (ARF). ARF is classified into prerenal, renal, and postrenal causes. GBM, Glomerular basement membrane; IgA, immunoglobulin A. (Adapted from Ashley J, et al: Pathophysiology and etiology of acute kidney injury. In Floege J, Johnson R, Freehally J, editors: Comprehensive clinical nephrology, ed 4, Philadelphia, PA, 2010, Mosby Elsevier, p 798.)
General Approach to Patients with Renal Dysfunction
Postoperative renal dysfunction is usually identified by oliguria or increases in serum creatinine. Evaluation of patients should include a thorough physical examination. Prerenal causes of renal dysfunction are most frequently encountered in the early postoperative period. Assessment of the patient’s intravascular volume status and cardiac performance is required. A patient with signs of volume depletion requires replenishment of intravascular volume with physiologic saline (without potassium supplement until renal failure is ruled out). If examination reveals that diminished cardiac performance is responsible for the oliguria, judicious inotropic support is provided while indices of cardiac performance are measured.29,30
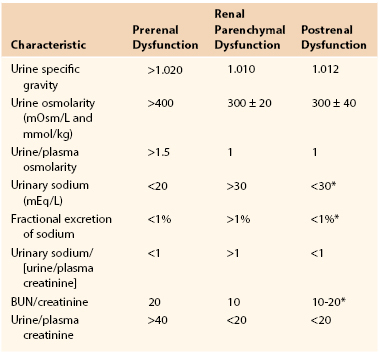
If correction of filling pressures or myocardial performance fails to improve urinary output, samples of urine and blood are obtained, and diuretic therapy is considered. Serum electrolytes, blood counts, and urine studies allow evaluation of other possible sources of oliguria and renal failure, such as ATN or myoglobinuria. Urine studies include urinalysis; urine sodium, urea, and creatinine concentrations; urine osmolality; and estimation of the fractional excretion of sodium (Table 41-1).
Acute Renal Dysfunction: Causes
Prerenal.
Prerenal causes are the most frequent source of acute renal dysfunction in the early postoperative period. Renal failure from a prerenal cause is usually the direct result of a contracted intravascular volume secondary to inadequate fluid replacement to replenish intraoperative and postoperative fluid losses. Less commonly, it is secondary to a primary reduction in cardiac performance that triggers neurohormonal reflexes to enhance intravascular volume by increasing tubular reabsorption of sodium and water. In their pure forms, these two causes of reduced renal function are easily discernible. Hypovolemia is associated with flat neck veins, dry mucous membranes, and reduced pulmonary artery wedge pressure, whereas renal dysfunction secondary to poor cardiac performance is associated with distended neck veins, clinical fluid overload, and elevated pulmonary artery wedge pressure. Therapy for hypovolemic prerenal azotemia is to increase intravascular volume by administering balanced salt solution and red blood cells as needed. Conversely, therapy for renal dysfunction of cardiogenic origin is directed at improving myocardial performance by administering afterload-reducing and inotropic agents, and instituting diuretic therapy as needed to diminish the preload of the failing left ventricle.
Because an atherosclerotic patient who is undergoing major vascular and endovascular procedures frequently has associated coronary artery disease and impaired left ventricular function, distinction between these two causes of prerenal dysfunction (hypovolemic vs cardiogenic) can be difficult. Preexisting heart disease may raise the baseline total body volume for an individual to higher central filling pressures, and apparently normal or low-normal cardiac filling pressures may reflect relative hypovolemia. In this clinical situation, one can maintain a constant infusion of afterload-reducing and inotropic agents (e.g., dobutamine) and cautiously administer small boluses of balanced salt solution while monitoring cardiac output and pulmonary artery wedge pressure. If no urinary response is noted when filling pressures begin to increase, diuretic therapy is added. The vascular surgeon should have reliable estimates of cardiac function and filling pressures before initiation of inotropic or diuretic therapy, or exacerbation of compromised renal function may result.31,32
Another important prerenal cause of acute and chronic renal insufficiency is occlusive disease of the renal arteries. Also termed ischemic nephropathy, this diagnosis is made by exclusion of other causes. If other causes of prerenal dysfunction have been excluded, renal duplex sonography is used to determine whether occlusive disease of the main renal arteries is present. Hemodynamically significant renal artery stenosis or occlusion is characterized by a focal increase in peak systolic velocity (>1.8 m/sec) along with distal turbulent Doppler waveforms (i.e., stenosis) or absence of Doppler flow (i.e., occlusion). ATN is characterized by a marked decrease in diastolic velocity and an increased resistance index measured by spectral analysis of the renal artery. In the absence of acute or chronic renal parenchymal disease, diastolic velocity is increased as a result of a compensatory decrease in renovascular resistance. When the duplex scan is positive and correction of renal arterial stenosis or occlusion is contemplated, contrast-enhanced angiography is performed to diagnose the problem, and the appropriate endovascular or operative intervention is instituted.33
Postrenal.
Postrenal mechanisms represent the least frequent cause of postoperative oliguria leading to renal dysfunction. The pathophysiology involved in this process is obstructive, and it is usually at the level of the urethra or urinary catheter and less commonly at the level of the ureters. Hematuria or traumatic catheter insertion can predispose to clots and obstruction of indwelling urinary catheters with resultant obstructive uropathy. For this reason, when rapid cessation of urine flow is detected, initial maneuvers should be directed toward catheter irrigation or replacement, which is usually successful in restoring urinary flow. Problems encountered with catheter insertion, such as the presence of clots or urethral strictures, should be noted. Similarly, catheter kinking should be avoided, because it can occasionally cause obstruction. Postrenal oliguria can also be caused by ureteral or renal pelvic obstruction, and these mechanisms should be investigated after other causes of oliguria have been excluded. Causes include iatrogenic injury or compression of the ureters associated with aortic surgery, graft placement, and stone disease. A preliminary diagnosis can be suggested on the basis of renal ultrasound or isotope renography, and can be confirmed with retrograde urography. Therapy may require the placement of ureteral stents or percutaneous nephrostomy to relieve the obstruction.29
Acute urinary retention from obstructive uropathy can rarely accompany removal of a urinary catheter. Clinical situations in which acute urinary retention can arise include voiding dysfunction after urinary catheter removal in patients with epidural catheters placed for pain control and in patients with prostatic hypertrophy. Generally, one should allow 6 to 12 hours to elapse after epidural analgesia is discontinued before removal of urinary catheters to avoid urinary retention. Prostatitis or traumatic urinary catheter insertion combined with general anesthesia can also precipitate acute urinary retention. Both these problems, if recognized early, are easily treatable by catheter reinsertion and rarely progress to obstructive uropathy.
Parenchymal.
Parenchymal causes of acute renal dysfunction are diverse and pose the greatest risk for permanent renal failure. ATN describes all renal parenchymal causes of ARF. More specifically, the pathophysiologic mechanism of ATN involves a decrease in cellular ATP, which is associated with loss of the actin cytoskeleton; loss of the cytoskeleton causes a loss of renal tubular cell membrane polarity and subsequent loss of intercellular tight junctions. Shedding of the apical portion of tubular cells into the tubules can result in tubular obstruction and lead to a further reduction or cessation of glomerular filtration in the nephron. ARF is manifested clinically by an abrupt rise in serum creatinine, either with or without a change in urinary output (oliguria); it is sometimes possible to detect the presence of tubular cells in the urinary sediment on microscopic urinary evaluation. Although ATN may be transient and self-limited, its causes related to vascular surgery include ischemic injury (shock, acute renal artery occlusion, multiorgan failure, and atheroembolic injury) and toxic injury (myoglobinuria and dye-related injury, see the section below (see Fig. 41-2).29,30
Ischemic Injury to the Kidney
Acute Ischemic Renal Injury
Postoperative acute ischemic renal injury is caused by either temporary periods of interruption of renal perfusion or periods of systemic hypoperfusion. The pathophysiology of acute ischemic injury is twofold. First, as a consequence of the magnitude and duration of ischemia, tubular cell swelling occurs after reperfusion. This swelling can cause tubular obstruction, leading to a further reduction or cessation of glomerular filtration in the nephron. Second, tubular cells either can lose their basement membrane attachment secondary to the interstitial edema that develops after reperfusion or can undergo cell death during ischemia, and subsequently, be sloughed into the tubule. The medullary thick ascending loop of Henle and the pars recta of the proximal tubule seem to be the segments of the tubular epithelium that are most sensitive to ischemia. After loss of the tubular cell, back leak of glomerular filtrate into the renal parenchyma develops.30–36 The risk for acute renal dysfunction after a vascular surgical intervention is greatest for aortic surgery and is described in more detail in the section titled Toxic Injury and Angiography.37–47
Acute renal artery occlusion is discussed thoroughly in Chapter 149. It can be caused by emboli from a cardiac origin, by trauma, or by aortic or renal artery dissection. When related to a cardioembolic event, the diagnosis is often delayed, and ultimate recovery of renal function depends on the magnitude of the occlusion and the presence of preexisting collaterals to the kidneys. Back, flank, or abdominal pain, new onset of hypertension, hematuria, and elevated serum lactate dehydrogenase levels may provide a clue regarding the diagnosis and treatment.48 Traumatic renal artery occlusion is suggested by the presence of hematuria and nonvisualization of the kidney on an intravenous pyelogram. Prompt angiography is necessary to confirm the process, although the success of revascularization largely depends on the duration of the ischemic period, which is frequently prolonged. Dissection of the renal artery can be due to catheter-related injury, or it can arise from preexisting disease (fibromuscular dysplasia). Treatment and opportunity for functional recovery are based on the extent of the dissection, and in cases of complete occlusion of the renal artery, the period of ischemia before surgical revascularization.
Atheroembolism to the renal arteries has increasingly been recognized as a cause of ARF and can give rise to renal damage culminating in end-stage renal disease (ESRD). Catheter-based peripheral and coronary angiography and endoluminal interventions are well-recognized sources of renal atheroemboli. Up to 20% to 40% of patients with renal artery interventions have shown deterioration in GFR. It would seem logical to include use of distal embolic protection during these procedures. This is supported by the study of Henry et al,49 which showed that 100% of the patients treated with renal artery angioplasty and stents with distal filter wire protection showed evidence of emboli on the filter. Cooper et al50 showed that, in addition to distal embolic protection, adjunct pharmacologic treatment (such as the platelet glycoprotein IIB/IIIa inhibitor, abciximab) may be required to minimize renal injury after endoluminal interventions. Atherosclerotic plaque from proximal diseased aortic segments can complicate suprarenal cross clamping or manipulation of the aorta. Atheroembolism can also occur spontaneously from these proximal sources or from renal artery atherosclerotic plaque. The clinical diagnosis is suggested by deterioration of renal function in a patient who displays other extrarenal manifestations of atheroembolism (e.g., blue toe syndrome) and is highly suggested by the laboratory finding of eosinophilia (71%). The diagnosis is confirmed by renal biopsy, and treatment is supportive.51–54
Vascular procedures complicated by sepsis, myocardial dysfunction, and reperfusion injury can result in transient or permanent renal dysfunction. In these instances, recovery of excretory renal function depends on elimination of the septic focus and improvement in left ventricular performance to ensure adequate renal perfusion.
Chronic Ischemia and Ischemic Nephropathy
Chronic ischemic nephropathy is discussed in Chapters 143 and 144. The term ischemic nephropathy describes reduced renal excretory function in conjunction with renovascular disease. Usually, the renovascular disease is bilateral or involves a solitary kidney, and diminished renal artery perfusion has been implicated as the cause of elevated serum creatinine. The significance of this condition is that it tends to be rapidly progressive and is thought to be responsible for up to 20% of these patients becoming dependent on dialysis. Uncorrected renovascular disease as a cause of ESRD is associated with a rapid rate of death during follow-up, with a median survival of only 27 months after the initiation of dialysis and a 5-year survival rate of just 12%. Patients who survive successful renal artery revascularization have an improved probability of long-term survival.55
In patients with atherosclerosis and recent worsening of renal function along with hypertension or worsening of renal function while taking an ACE inhibitor, the presence of ischemic nephropathy should be investigated. In contrast to renovascular hypertension secondary to unilateral renal artery stenosis, in which hypertension is renin dependent, hypertension in ischemic nephropathy tends to be volume dependent. Patients with this pattern of disease exhibit severe hypertension, elevated serum creatinine, and volume overload. Alternatively, the clinical manifestation may be recurrent episodes of flash pulmonary edema. This entity almost always coexists with some element of intrinsic renal parenchymal disease.56–59
Toxic Injury and Angiography
Chemical injury to the kidney can result from many sources (Fig. 41-3). Nephrotoxic agents should be identified and used with caution, particularly in a vascular patient with compromised renal clearance. Aminoglycosides are some of the most common compounds responsible for such injury in the postoperative period; myoglobin and radiologic contrast media have also been implicated. Aminoglycosides appear to exert their renal toxicity at the tubular cell by causing mitochondrial damage, destruction of the cell membrane, activation of phospholipase, or alteration in lysosomes. Because of this relationship and the frequent history of reduced renal function in postoperative vascular surgery patients, it is important to identify risk factors that can contribute to nephrotoxicity before the administration of aminoglycosides. Such risk factors include preexisting renal insufficiency, advanced age, extracellular volume depletion, and concomitant use of other nephrotoxins. The routine use of aminoglycoside blood levels to predict or prevent nephrotoxicity is probably not warranted. Although alternative antibiotics with less nephrotoxicity have reduced the use of aminoglycosides in vascular surgical patients, all pharmacologic agents should be administered with caution, and the dosage adjusted for renal clearance.60–62
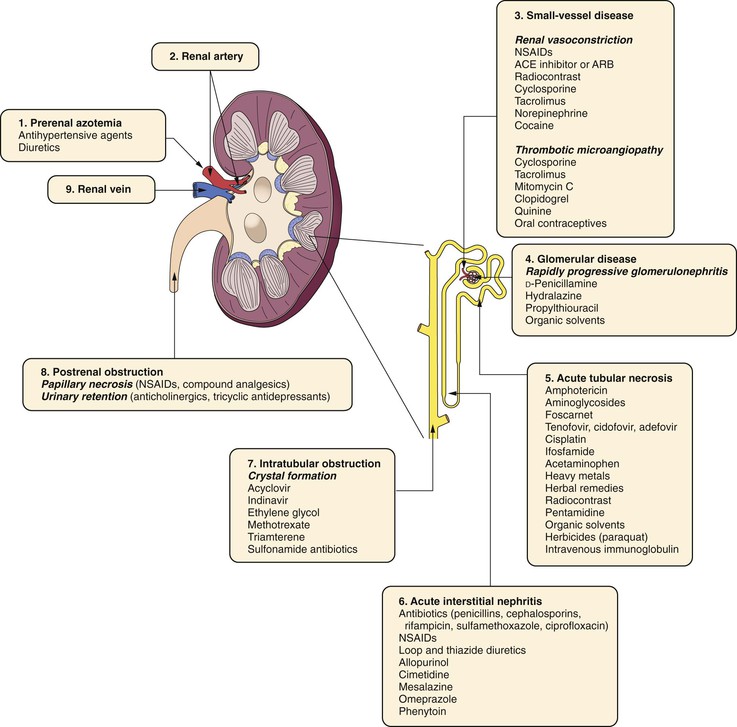
Figure 41-3 Nephrotoxic agents that lead to acute renal failure. ACE, Angiotensin-converting enzyme; ARB, angiotensin receptor blocker; NSAID, nonsteroidal anti-inflammatory drug. (Adapted fromAshley J, et al: Pathophysiology and etiology of acute kidney disease. In Floege J, Johnson R, Freehally J, editors: Comprehensive clinical nephrology, ed 4, Philadelphia, PA, 2010, Mosby Elsevier, p 804.)
Myoglobinuria is an important cause of renal failure in patients who undergo revascularization after prolonged periods of limb ischemia. Circulating as a breakdown product of muscle death, myoglobin is filtered freely by the glomerulus. Myoglobin exerts its toxicity through direct tubular cell injury and via precipitation and obstruction of the tubule. Hematuria after reperfusion of a profoundly ischemic extremity suggests pigment toxicity and should prompt urinalysis. Myoglobinuria is suggested when the urine is dipstick positive for blood but no red blood cells are present on microscopic analysis, and it can be confirmed by testing the urine for myoglobin. When diagnosed, injury to the kidney may be lessened by maximizing the urine flow rate through the infusion of intravenous crystalloid and diuretics (mannitol) and by alkalinizing the urine (sodium bicarbonate).63
Administration of contrast media continues to be a frequent cause of hospital-acquired renal failure. Over the past few years, the increasing numbers of diagnostic and interventional procedures requiring contrast agents has paralleled the increasing incidence of contrast media–induced nephropathy (CMIN). CMIN now accounts for more than 10% of cases of hospital-acquired renal failure and is the third leading cause of iatrogenic ARF in hospitalized patients. The increased incidence of CMIN is associated with an increase in hospital mortality, length of stay, and cost.64–73
Conventional contrast agents have iodine incorporated into their structure to absorb x-ray photons, thereby achieving visualization of the vasculature. The nephrotoxicity of such iodinated contrast agents has been recognized for many years. The principal site of contrast-induced nephrotoxicity is the renal tubule from transient regional renal ischemia; the effects on glomerular function seem to be mild.74 Nonionic contrast agents are now available and provide comparable absorption of x-ray photons, yet are significantly less charged than traditional agents. However, severe adverse renal events still continue to plague the use of nonionic contrast media.71
Renal nephrotoxicity after exposure to ionic agents occurs most commonly in patients with preexisting renal insufficiency (relative risk of 3.3) alone or in combination with diabetes mellitus, especially diabetes type 1. Other risk factors, such as dehydration, volume of contrast agent used, and simultaneous exposure to other nephrotoxins, contribute to the likelihood of acute contrast-induced nephrotoxicity.70 Additional risk factors include multiple myeloma and heavy proteinuria. Overall, the incidence of acute renal dysfunction after contrast-enhanced angiography varies from 0% to 10%, although these estimates are skewed by several studies that included only diabetes type 1 patients. In one study, hospital-acquired nephropathy occurred in 12% of patients. In patients with normal renal function, however, the incidence of CMIN is just 1% to 2%.71 Although diabetic and nondiabetic patients with renal insufficiency are at increased risk for contrast-induced ARF, diabetics seem to recover less often and are at greater risk for permanent dependence on dialysis as a consequence of contrast-induced ARF.74–79
One of the difficulties in treating or preventing renal failure after the administration of contrast media is that the biochemical mechanisms of CMIN have not been identified. One group of studies has hypothesized that NO contributes to preservation of renal blood flow and function in normal and pathologic states. Several studies have shown that inhibition of NO, before the administration of radiocontrast material, results in abolition of the medullary vasodilatory response and contributes to regional hypoxic renal tubular injury.80–83 Heyman et al81 also suggested that maintenance of local vasodilatory prostaglandins, in particular PGE2, is likewise important in maintaining regional blood flow to the medulla. Myers et al84,85 recently combined the use of in vivo microdialysis and laser Doppler blood flow analysis to identify the clinically relevant intrarenal vasodilators (cortical and medullary) that are required to maintain microvascular blood flow during exposure to contrast material. Conray (Mallinckrodt, Hazelwood, Mo), an ionic contrast agent, caused a marked decrease in cortical and medullary blood flow with a concomitant decrease in endogenous cortical NO and PGE2 and medullary NO synthesis. Conray did not significantly alter the content of inducible nitric oxide synthase (iNOS), COX-2, prostacyclin synthase, or PGE2 synthase. These findings suggest that downregulation of renal cortical and medullary NO synthesis contributes to the contrast-induced loss of renal cortical and medullary microvascular blood flow. They also suggest that preservation of normal levels of renal cortical and medullary NO synthesis may help prevent or lessen the contrast-induced renal injury that occurs after diagnostic and therapeutic endovascular procedures.86
As stated earlier, nonionic contrast agents were developed in the expectation that they would be less toxic to the kidney. However, these agents continue to cause CMIN, but the mechanisms of this injury are unknown. Myers et al84–86 used their experimental model, described earlier, to determine whether the mechanisms of nonionic CMIN are different or similar to those of ionic CMIN. The ionic agent Conray decreased superior mesenteric artery (SMA) blood flow, whereas the nonionic agent Optiray (Mallinckrodt) did not. Neither Conray or Optiray altered total renal or aortic blood flow. Conray caused a marked decrease in cortical and medullary blood flow and NO synthesis, but did not alter PGI2 or PGE2. Optiray decreased medullary and cortical blood flow and cortical NO synthesis. Unlike Conray, Optiray did not decrease medullary NO synthesis, but profoundly decreased medullary and cortical PGE2 and PGI2 synthesis. These findings suggest that both Conray (ionic contrast agent) and Optiray (nonionic contrast agent) decrease renal cortical and medullary blood flow and renal function, although the intrarenal mechanisms differ. The decrease in renal cortical and medullary blood flow occurred without a decrease in total renal blood flow, thus suggesting that the decrease in renal microvascular blood flow induced by ionic and nonionic contrast agents occurs as an early injury. The Conray-induced fall in SMA blood flow may contribute to intestinal ischemia-reperfusion, which can contribute to distant organ dysfunction, including the kidney. This finding suggests that ionic contrast–induced renal injury is due to local and systemic mechanisms. One must consider that preservation of normal levels of renal cortical and medullary NO synthesis and SMA blood flow may be required to prevent or lessen ionic and nonionic contrast–induced renal injury.87
Specific measures to minimize the risk for contrast-induced ARF are controversial, and the results from controlled studies are largely inconclusive. Nevertheless, the basic relationship between the use of contrast material and the risk for contrast-induced nephropathy seems to be related to the amount of time that the kidney is exposed to the contrast material. For this reason, maximizing the urine flow rate during and immediately after angiography and limiting the quantity of contrast agent used are important considerations. Maximal urine flow rates should be achieved by preliminary intravenous hydration of the patient. Studies investigating the optimal preparation of patients with renal insufficiency indicated that hydration with 0.45% saline provides better protection against the acute decline in renal function associated with radiocontrast agents than does hydration with 0.45% saline plus mannitol or furosemide.88 One regimen treats any patient with the aforementioned risk factors 12 hours before angiography with intravenous hydration at 1.5 mL/kg/hr. Immediately before angiography, the patient usually receives a bolus of intravenous fluid (3 to 5 mL/kg). Finally, intravenous hydration is continued for 4 to 6 hours after completion of the study. Another approach to hydration was presented by Merten et al,89 who demonstrated the potential efficacy of sodium bicarbonate hydration before, during, and after the use of contrast agents. In this regimen, patients are treated with 154 mEq/L of sodium bicarbonate with a bolus of 3 mL/kg, followed by 2 mL/kg/hr for 6 hours after the procedure. We have used this protocol with very few instances of contrast-induced renal injury over a 4-year period.
Although attempts to calculate a safe upper limit of contrast material have met with some success, no definitive limit currently exists. Even small doses (30-60 mL) may induce renal failure in patients with extreme renal insufficiency (GFR ≤15 mL/min). Conversely, more than 300 mL of contrast material may be administered safely to other patients with no risk factors for ARF.90 It seems prudent to limit the quantity of nonionized contrast agent to less than 50 to 75 mL in patients with a significant reduction in GFR (<20 to 30 mL/min). In some instances, digital subtraction techniques have been useful in limiting the quantity of contrast material required. Nevertheless, a single midstream aortic injection of 30 to 40 mL of nonionic contrast material with conventional cut-film techniques is just as safe as digital subtraction angiography.
Adjuncts or alternatives to conventional angiography are appropriate in many instances. In addition to the use of digital subtraction techniques, carbon dioxide gas can be used for angiography with minimal renal risk.91 Because it offers limited detail, carbon dioxide angiography is often used to identify the site of disease, and then it is better defined with conventional contrast agents. Another alternative to conventional angiography that eliminates the risk of nephrotoxicity is the use of abdominal ultrasound with visceral and/or renal artery duplex sonography. Use of magnetic resonance angiography with gadolinium-based contrast agents is no longer considered a viable option in these cases because of reports of nephrogenic fibrosis in patients with compromised renal function.92
By scavenging reactive oxygen species, acetylcysteine may protect against contrast-induced nephrotoxicity. Tepel et al93 studied patients with chronic renal dysfunction who required administration of nonionic contrast for computed tomography. They documented a significant reduction in serum creatinine with the use of oral acetylcysteine and hydration versus placebo and hydration.93 However, there are as many studies showing either a benefit or no benefit from the use of acetylcysteine.93–96 One regimen describes the administration of two oral doses of acetylcysteine (600 mg) before and after these studies in patients at high risk for CMIN. Finally, high-dose loop diuretics, ACE inhibitors, and angiotensin II receptor antagonists are withheld for at least 72 hours before aortic reconstruction or exposure to arterial contrast agents. Selective beta blockers and calcium channel blockers are substituted when necessary.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


