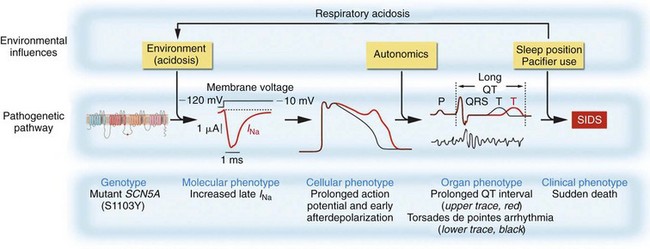
98 Linking Sudden Infant Death Syndrome and Arrhythmia Sudden Infant Death Syndrome and Long QT Syndrome Sudden Infant Death Syndrome and Other Arrhythmia Syndromes Congenital Arrhythmia and Sudden Infant Death Syndrome: How Frequent? Congenital Arrhythmia in Sudden Infant Death Syndrome: Triple Risk Hypothesis Sudden infant death syndrome (SIDS) is defined as the sudden, unexpected death of an infant (before the age of 1 year) for which no cause is apparent.1 Sudden unexpected death syndrome refers to the unexpected death of a person older than 1 year but younger than 35 or 40 years.2 This chapter addresses only SIDS, but the age demarcation of 1 year is somewhat arbitrary, and overlap surely exists for the two syndromes. Depending on the precise definition of SIDS, the historical era, demographics, and ethnic status of the population, SIDS affects approximately 0.1 to 2 infants per 1000 live births, with a peak incidence between the ages of 2 and 5 months.1,2 The etiologic basis for SIDS can be neurologic, endocrine, metabolic, pulmonary, immune, or cardiac with a genetic basis implicated for each.3 This chapter reviews the evidence that cardiac arrhythmia has a role in SIDS, with a focus on genetic predisposition to fatal arrhythmia.3 The literature on SIDS in general, and on the role for arrhythmia and SIDS in particular, is vast, with numerous studies, reviews, editorials, and commentaries highlighting the controversies. The focus of this chapter is on more recent studies and review articles that will lead the interested reader to this rich literature. By definition, SIDS occurs in infants without diagnosed or suspected disease antemortem, and with no clear cause of death on routine postmortem examination. This lack of antemortem data has been an obstacle for the field overall and particularly for making the link to arrhythmia. Techniques to investigate the etiology of SIDS include analyses of epidemiologic data, studies of the parents and relatives of SIDS victims, and studies of infants who were considered to be at high risk for SIDS. Infants who came to medical attention before death, or who were resuscitated from an event (called survivors of SIDS, or designated as having near miss SIDS) also have been studied. These cases provide valuable insights into and proof of principle for etiologic factors in SIDS. The near-miss cases, however, do not fit the clinical and epidemiologic definition of SIDS, because the affected patients either had clinical manifestations detected antemortem or did not die. With the discovery that mutations in ion channels and associated proteins cause long QT syndrome (LQTS; Chapters 50 and 69) and other arrhythmia syndromes such as Brugada syndrome (BrS; Chapters 50 and 92) and catecholaminergic polymorphic ventricular tachycardia (CPVT; Chapter 53), it became possible to perform a “molecular autopsy”2 for SIDS victims by screening candidate genes for mutations. It also is important to recognize that the presence of a mutation in a susceptible gene does not necessarily imply a link to causation, because mutations without functional significance can occur as common polymorphisms (affecting greater than 0.5% of the population) or as rare variants (affecting less than 0.5% of the population). The absence of the mutation in control panels provides supporting evidence for pathogenicity, but the possibility of discovering an incidental rare variant in SIDS increases as the number of genes screened increases. Traditional linkage studies in family kindreds are used to connect the genotype to the phenotype, but such studies are not possible in SIDS because the phenotype is death in infancy, and mutations can be de novo and are not present in the parents. When a mutation is found, biophysical function of the mutation can be investigated by expressing the mutated protein in heterologous expression systems, or even in myocytes, to demonstrate an abnormality consistent with known pathogenetic mechanisms for LQTS, BrS, CPVT, or other arrhythmia syndromes. If such a mutation is present, it provides strong but not conclusive evidence for pathogenicity. If it is absent, however, it does not prove nonpathogenicity because the mutation might not have been studied under the conditions necessary to elicit the dysfunction (e.g., reduced pH),4,5 or that the presence of the complete macromolecular complex present in myocytes but not heterologous cells is required to manifest dysfunction. To support the conclusion of a link between a genotype and an arrhythmic SIDS clinical phenotype, a mutation should cause biophysical dysfunction of an ion current under relevant environmental conditions that result in a cellular phenotype supporting an arrhythmogenic phenotype at the tissue and organ level (Figure 98-1). Figure 98-1 The link between genotype and phenotype in sudden infant death syndrome (SIDS) emphasizing multiple levels of phenotype and interaction with the environment. A mutation in the cardiac sodium channel gene SCN5A is associated with a pathologic molecular (also called biophysical) phenotypic feature of increased late sodium current, which in turn results in an abnormal cellular phenotype, in this case action potential prolongation and afterdepolarizations, which causes tissue or organ manifestations of prolonged QT interval and arrhythmia, eventuating in the clinical phenotype of SIDS. (From Makielski JC: SIDS: Genetic and environmental influences may cause arrhythmia in this silent killer. J Clin Invest 116:297, 2006.) In the 1970s, Schwartz6 suggested that abnormalities of cardiac sympathetic innervation caused a disorder of the heart’s electrical rhythm known as LQTS, and that this can also cause SIDS.6 Maron et al.7 showed a longer QT interval in parents of SIDS victims and provided evidence for a role of LQTS in SIDS. In a prospective trial of electrocardiographic (ECG) screening in more than 34,000 infants that produced 24 SIDS cases,8 a long QT interval increased the risk of SIDS by a factor of 41. The proof of principle that congenital LQTS could cause SIDS came from two cases: a near-miss case9 in which the infant survived, and a case in which the patient died in infancy but LQTS was recognized antemortem.10 In both cases, a mutation in the gene SCN5A encoding the voltage-dependent cardiac sodium channel was found, and it had the characteristic biophysical phenotype of increased late sodium current associated with LQTS type 3 (LQT3). These cases, however, did not meet the formal epidemiologic definition of SIDS. In 2001, Ackerman et al.11 screened a cohort of 93 patients with defined SIDS for mutations in SCN5A and found two novel mutations that on expression in heterologous cells showed the increased late sodium current of LQT3. Additional studies in SIDS cohorts have found additional mutations in SCN5A associated with LQT3.4,12 Most of these mutations were rare and novel, but one, S1103Y, was a common polymorphism with an allelic frequency of approximately 10% in the black population that had previously been shown to be associated with ventricular arrhythmia in adults.13 Plant et al.4 showed that homozygosity for S1103Y increased the risk of SIDS by 24-fold. Moreover, the biophysical abnormality of increased late sodium current depended on acidosis, linking the interaction of environmental influences with the genotype to produce the pathologic phenotype (see Figure 98-1). Subsequently, two other mutations were shown to require acidosis to produce the abnormal phenotype.5 Three novel mutations in caveolin-3 (encoded by CAV3), a channel accessory protein, were found in a SIDS cohort and were shown to cause late sodium current characteristic of LQTS.14 Four novel mutations in α-syntrophin (encoded by SNTA1), a scaffolding protein associated with SCN5A, were found in a SIDS cohort and were shown to cause increased late sodium current by a nitrosylation dependent mechanism.15 Novel mutations in the sodium channel–associated β-subunits β3 (encoded by SCNB3) and β4 (encoded by SCNB4) were found in the same SIDS cohort and shown to cause increased late sodium current.16 Thus, the abnormality of sodium current in LQTS as a cause of SIDS is not restricted to mutations in SCN5A. Mutations in potassium channel genes KCNQ117 (LQT1), KCNH18 (LQT2), and KCNE12 (LQT6), which cause loss of function in potassium currents, have also been described in SIDS cohorts. The common KCNH2 polymorphism K897T was found to be strongly associated with two cases of SIDS in a singular family.19 Two other LQTS potassium channel genes, KCNJ2 (LQT7) and KCNE1 (LQT5), were screened in 201 SIDS cases,12 but no pathologic mutations were found. Currently, approximately 80% of all genetic LQTS can be accounted for by mutations in the known genes (for LQT1 to LQT13),20
Sudden Infant Death Syndrome
Linking Sudden Infant Death Syndrome and Arrhythmia
Sudden Infant Death Syndrome and Long QT Syndrome
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thoracic Key
Fastest Thoracic Insight Engine
