CHAPTER 4
Structural Heart Disease and Progression to Failure: Stages B, C, and D
“DNA neither cares nor knows. DNA just is.
And we dance to its music.”
—Richard Dawkins,
River Out of Eden: A Darwinian View of Life1
Morphologic Changes in Heart Failure
Initiation and progression of disease is the product of genetic predisposition, environment, and chance. Following a primary injury to the heart, altered mechanical loading and neurohumoral signals orchestrate a secondary transition in cardiovascular phenotype. In part, this manifests as a reinitiation of a fetal repertoire of gene expression. Over time, qualitative and quantitative changes in proteins regulating myocyte contractility and calcium homeostasis contribute to impaired systolic and diastolic ventricular function.
HEART SIZE
Initial structural heart damage progresses to chronic heart failure associated with increases in heart mass and size (Figure 4.1). Although this progression is mostly due to increases in preexisting myocyte size, proliferation of resident endogenous and circulating exogenous stem cells may contribute to this process.2 The different phenotypes of hypertrophy depend on the inciting factors for growth (Figure 4.2). Patients with HF-pEF, compared to normal controls and hypertensive patients without heart failure, exhibit an average 33% increase in left ventricular mass and wall thickness, but without change in end-diastolic chamber volume.3 Patients with HF-rEF, compared to controls, demonstrate more than twofold increases in left ventricular mass and end-diastolic chamber volume.4
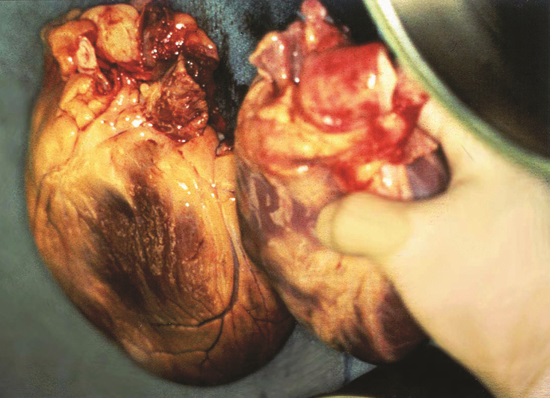
FIGURE 4.1 Heart size. Markedly enlarged heart removed from patient with end-stage heart failure due to ischemic cardiomyopathy (left) versus normal sized donor heart about to be transplanted (right).

FIGURE 4.2 Common pathologies of heart failure remodeling. Postmortem examples: Panel A: Hypertensive hypertrophic cardiomyopathy. Panel B: Dilated ischemic cardiomyopathy. Panel C: Dilated nonischemic cardiomyopathy. The pathology shown in Panel A is associated with HF-pEF, whereas those in Panels B and C are associated with HF-rEF.5 Source: Adapted from Konstam, J Card Fail. 2003;9(1):1-3, with permission.
VENTRICULAR REMODELING
The heart is often viewed as a structurally stable, mechanical tissue pump that adjusts its performance in response to changes in loading conditions or inotropic state. When viewed by electron microscopy, repeating sarcomere units appear to be permanent, crystalline structures. In fact, the heart is a dynamic biologic structure with ongoing turnover of its intracellular contracting components associated with the continuous assembly and degradation of specific sarcomere proteins. On average, troponin subunits (T/I/C) have a half-life of 3 to 5 days; actin and tropomyosin 7 to 10 days; and myosin 5 to 8 days.6 Thus, the heart, while cyclically generating force and motion, is an organ with potential for great plasticity that is primed for cellular and structural modulation when changes in mechanical and molecular signals occur.
Myocardial injury alters the loading and biochemical environment of both impaired and uninjured cardiac cells. Biochemical signals, whether endocrine (coming from an external source), paracrine (acting on neighboring cells), autocrine (acting on the same cell), or intracrine (acting internally on the same cell without extracellular secretion), can all contribute to a subsequent net biologic response.7 As in other tissues, these mediators in part reinitiate fetal growth repertoires of transcription and translation.8 Over time, characteristic patterns of heart morphology emerge associated with different effects on ventricular filling or ejection of blood (Figure 4.2).
Cell proliferation, apoptosis, hypertrophy, and atrophy modify the physical characteristics of the heart—a process referred to as ventricular remodeling.8 While remodeling may initially represent a reparative response to abnormal myocardial conditions, if sustained, it can ultimately contribute to ventricular dysfunction. This paradox may in part be explained by a limited set of mechanisms in response to transient cellular stress that have not evolved in the face of the current major causes of persistent adult cardiac disability, such as coronary artery disease and hypertension.
Patterns of Maladaptive Hypertrophy
Ventricular dysfunction occurs in part because of maladaptive hypertrophy. The loading conditions on the left ventricle and the extracellular environment both contribute to the processes of remodeling that result in two main classifications of hypertrophy.
CONCENTRIC VERSUS ECCENTRIC HYPERTROPHY
The normal left ventricle surrounds a rotated ellipse or football-shaped chamber with end-diastolic wall thickness ≤ 11 mm and chamber diameter ≤ 56 mm. During systolic wall thickening and ejection of blood, the base of the left ventricle, including the mitral and aortic valves, moves toward a relatively stationary apex.9 The return motion of the base during early ventricular filling can be measure by echocardiography to assess diastolic function (see Chapter 7, Figure 7.3).
Concentric Hypertrophy
Concentric hypertrophy can develop in response to a sustained increase in left ventricular systolic pressure (e.g., hypertension or aortic stenosis). In response, end-diastolic wall thickness and relative wall thickness (wall thickness/chamber dimension) increase.10 This increase in wall thickness by the Laplace relation results in a decrease in afterload wall stress (pressure × radius/wall thickness) at end-systole, which means that ejection fraction and stroke volume may initially be maintained. A left ventricle subjected to long-standing pressure overload, however, can progress to a dilated ventricle with eccentric hypertrophy, reduced ejection fraction, and decreased stroke volume (Figure 4.3).
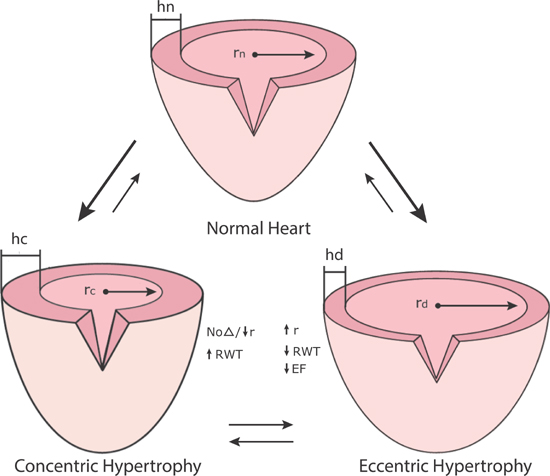
FIGURE 4.3 Concentric and eccentric hypertrophy. Abbreviations: r, chamber radius; h, wall thickness; σ, wall stress (force/area of muscle); EF, ejection fraction; RWT, relative wall thickness.11 Source: Adapted with permission from Jaski BE, Basics of Heart Failure. Springer Science + Business Media B.V.; 2000: 61.
Eccentric Hypertrophy (Dilated Left Ventricle)
Eccentric hypertrophy can follow myocardial injury (e.g., infarction, genetic cardiomyopathy, or myocarditis), a chronic volume overload (e.g., aortic or mitral valve regurgitation), or a decompensated state of concentric hypertrophy (Figure 4.3). For example, initially after myocardial infarction, passive stretch will help maintain a forward stroke volume via the Frank-Starling mechanism. Thinning and elongation of the fibrous infarcted segment further increase heart size (Figure 4.4).10 Eventually, increased wall stress and neurohumoral activation effect ventricular remodeling of the infarcted and noninfarcted myocardium, increasing chamber size, decreasing relative wall thickness, and decreasing ejection fraction.
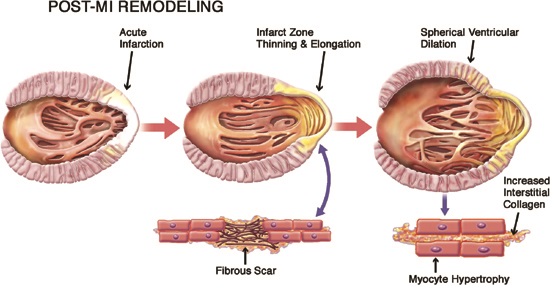
FIGURE 4.4 Post-MI left ventricular remodeling. Schematic representation of post-myocardial infarction (post-MI) left ventricular remodeling. The early phase (middle frame) is characterized by thinning and elongation of the fibrous scar within the infarcted zone. Subsequent left ventricular dilation, with transition from an elliptical to a more spherical configuration (final frame), is driven by neurohumoral activation and increased systolic wall stress resulting in diffuse myocyte hypertrophy associated with increased apoptosis (not shown) and increases in interstitial collagen.10 Source: Adapted with permission from Konstam et al., JACC: Cardiovasc Imaging. 2011;4(1):98-108.
LOADING CONDITIONS AND MYOCYTE PHENOTYPE
In 1975, Grossman and coworkers proposed the hypothesis that increased wall stress in either systole or diastole initiates either concentric or eccentric (dilated) hypertrophy (Figure 4.5).12 Since then, the molecular mechanisms for components of this control circuit have been identified. One mechanism involves a deformable sarcomere protein that undergoes differential phosphorylation based on the direction of stress and strain.13 Other molecular signals for the development of hypertrophy include stretch of the sarcomere-spanning protein titin and release of locally acting growth factors.14
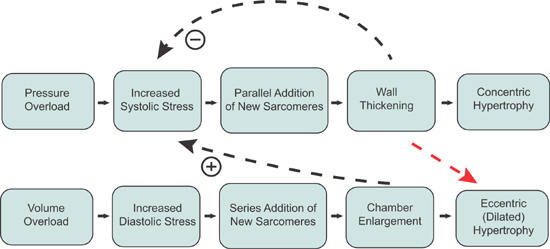
FIGURE 4.5 Loading conditions and sarcomere growth patterns. Hypothesized mechanism by which increased wall stress initiates concentric versus eccentric (dilated) hypertrophy. This hypothesis requires that the heart can “detect” systolic and diastolic wall stress occurring with pressure and volume overload, respectively, resulting in the selective addition of new sarcomeres in either a parallel or series position to return wall stress toward normal. If pressure overload is sustained, it can eventually also lead to eccentric hypertrophy (red arrow).12 Source: Adapted with permission from Grossman W et al., J Clin Invest. 1975;56(1):56-64.
EXTRACELLULAR ENVIRONMENT
The extracellular environment provides scaffolding for and participates in differentiation of cardiac muscle and nonmuscle cells.15 In parallel with changes of intracellular components, extracellular matrix modifications16 also participate in ventricular remodeling (Figure 4.6). Following ischemic or nonischemic insults, increased deposition of fibrous proteins, including collagen17 and fibronectin,18 further modulate the mechanical properties of the remodeled left ventricle.
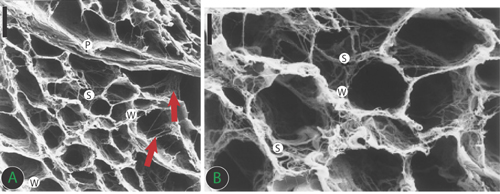
FIGURE 4.6 Connective tissue skeleton of human heart with an organization similar to a honeycomb. The perimysium (P) envelops groups of myocytes. The endomysial weave (W), as final arborization of the perimysium, supports and connects individual cells and is connected to adjacent myocytes by lateral struts (S). Collagen struts also connect myocytes to interstitial microvessels (lower arrow) or perimysium (upper arrow). Panel A: Low magnification ×1415, scale bar = 20 μm; Panel B: High magnification × 2830, scale bar = 10 μm.19 Source: Adapted with permission from Rossi MA et al., Circulation. 1998;97(9):934-935.
Neurohumoral Circulatory Responses
Complex neurohumoral responses amplify maladaptive remodeling and circulatory changes initiated by any insult to the heart.
ACUTE VERSUS CHRONIC NEUROHUMORAL RESPONSES
Regulatory mechanisms of sodium retention and vasoconstriction are appropriate to maintain homeostasis in response to acute hypoperfusion and hypotension following reductions in circulating blood volume such as hemorrhage (Table 4.1). These same responses, however, may be inappropriate if initiated in response to the development of chronic cardiac dysfunction and may contribute to further deterioration, thus becoming targets of therapy.20
TABLE 4.1 Neurohumoral responses to impaired cardiac performance and their effects on the circulation.20
RESPONSE | SHORT-TERM EFFECTS | LONG-TERM EFFECTS |
SALT AND WATER RETENTION | Augments preload | Pulmonary congestion, edema/anasarca |
VASOCONSTRICTION | Maintains blood pressure for perfusion of vital organs (brain, heart) | Exacerbates pump dysfunction (excessive afterload, increases cardiac energy expenditure) |
SYMPATHETIC STIMULATION | Increases heart rate and contractility | Increases energy expenditure |
NEUROHUMORAL MEDIATORS OF ADVERSE CARDIAC REMODELING
Following a primary myocardial injury, such as acute myocardial infarction or viral myocarditis, the onset of symptoms of pump failure may occur immediately, follow days of fluid retention, or follow months to years of maladaptive ventricular remodeling. The same neurohumoral mediators that act as vasoconstrictors or mediators of sodium and water retention can also act as growth factors that promote the progression of adverse ventricular hypertrophy, if sustained (Figure 4.7).
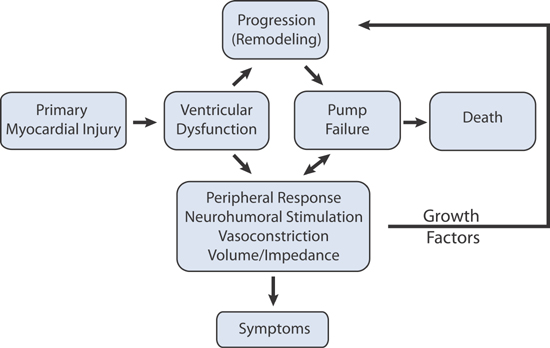
FIGURE 4.7 The Adverse Cardiac Remodeling Cascade. Effects of neurohormones and cytokine stimulation on the heart and the systemic vasculature in heart failure.11 Source: Adapted with permission from Jaski BE, Basics of Heart Failure. Springer Science + Business Media B.V.; 2000: 45.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


