Chapter 49 Stomach
Anatomy
Gross Anatomy
Divisions
The stomach begins as a dilation in the tubular embryonic foregut during the fifth week of gestation. By the seventh week, it descends, rotates, and further dilates with a disproportionate elongation of the greater curvature into its normal anatomic shape and position. Following birth, it is the most proximal abdominal organ of the alimentary tract. The most proximal region of the stomach is called the cardia, which attaches to the esophagus. Immediately proximal to the cardia is a physiologically competent lower esophageal sphincter. Distally, the pylorus connects the distal stomach (antrum) to the proximal duodenum. Although the stomach is fixed at the gastroesophageal (GE) junction and pylorus, its large midportion is mobile. The fundus represents the superiormost part of the stomach and is floppy and distensible. The stomach is bounded superiorly by the diaphragm and laterally by the spleen. The body of the stomach represents the largest portion and is also referred to as the corpus. The body also contains most of the parietal cells and is bounded on the right by the relatively straight lesser curvature and on the left by the longer greater curvature. At the angularis incisura, the lesser curvature abruptly angles to the right. It is here that the body of the stomach ends and the antrum begins. Another important anatomic angle (angle of His) is that formed by the fundus with the left margin of the esophagus (Fig. 49-1).
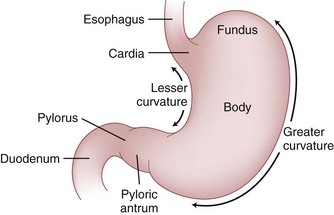
FIGURE 49-1 Divisions of the stomach.
(From Yeo C: Shackelford’s surgery of the alimentary tract, ed 6, Philadelphia, 2007, WB Saunders.)
Blood Supply
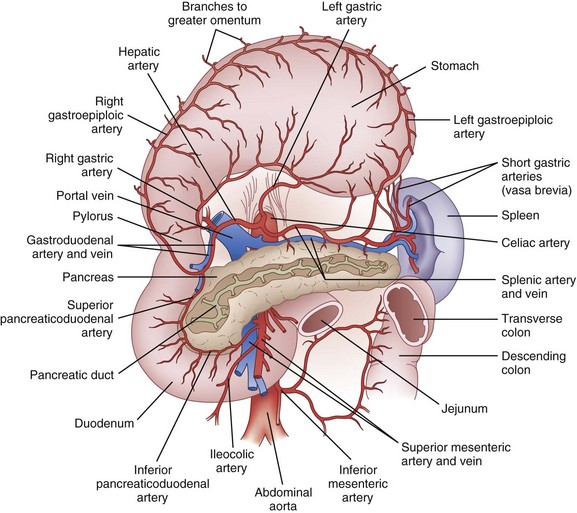
The celiac artery provides most of the blood supply to the stomach (Fig. 49-2). There are four main arteries—the left and right gastric arteries along the lesser curvature and the left and right gastroepiploic arteries along the greater curvature. In addition, a substantial quantity of blood may be supplied to the proximal stomach by the inferior phrenic arteries and by the short gastric arteries from the spleen. The largest artery to the stomach is the left gastric artery, and it is not uncommon (15% to 20%) for an aberrant left hepatic artery to originate from it. Consequently, proximal ligation of the left gastric artery occasionally results in acute left-sided hepatic ischemia. The right gastric artery arises from the hepatic artery (or the gastroduodenal artery). The left gastroepiploic artery originates from the splenic artery and the right gastroepiploic originates from the gastroduodenal artery. The extensive anastomotic connection between these major vessels ensures that in most cases, the stomach will survive if three out of four arteries are ligated, provided that the arcades along the greater and lesser curvatures are not disturbed. In general, the veins of the stomach parallel the arteries. The left gastric (coronary) and right gastric veins usually drain into the portal vein. The right gastroepiploic vein drains into the superior mesenteric vein and the left gastroepiploic vein drains into the splenic vein.
Lymphatic Drainage
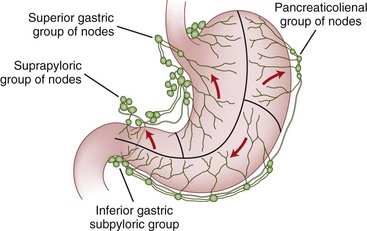
The lymphatic drainage of the stomach parallels the vasculature and drains into four zones of lymph nodes (Fig. 49-3). The superior gastric group drains lymph from the upper lesser curvature into the left gastric and paracardial nodes. The suprapyloric group of nodes drains the antral segment on the lesser curvature of the stomach into the right suprapancreatic nodes. The pancreaticolienal group of nodes drains lymph high on the greater curvature into the left gastroepiploic and splenic nodes. The inferior gastric and subpyloric group of nodes drains lymph along the right gastroepiploic vascular pedicle. All four zones of lymph nodes drain into the celiac group and into the thoracic duct. Although these lymph nodes drain different areas of the stomach, gastric cancers may metastasize to any of the four nodal groups, regardless of the cancer location. In addition, the extensive submucosal plexus of lymphatics accounts for the fact that there is frequently microscopic evidence of malignant cells several centimeters from gross disease.
Innervation
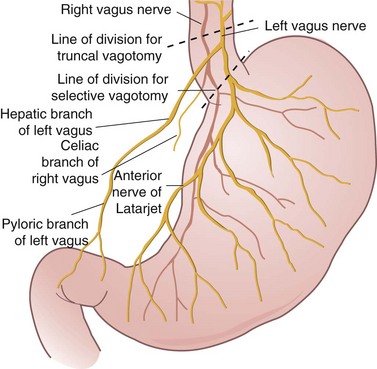
As shown in Figure 49-4, the extrinsic innervation of the stomach is parasympathetic (via the vagus) and sympathetic (via the celiac plexus). The vagus nerve originates in the vagal nucleus in the floor of the fourth ventricle and traverses the neck in the carotid sheath to enter the mediastinum, where it divides into several branches around the esophagus. These branches coalesce above the esophageal hiatus to form the left and right vagus nerves. It is not uncommon to find more than two vagal trunks at the distal esophagus. At the GE junction, the left vagus is anterior, and the right vagus is posterior (LARP).
Gastric Microscopic Anatomy
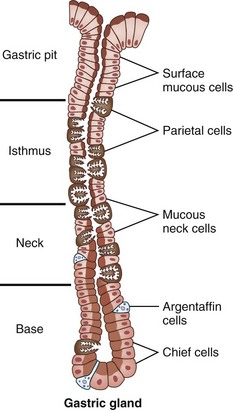
Gastric mucosa consists of columnar glandular epithelia. The cellular populations (and functions) of the cells forming this glandular epithelium vary based on their location in the stomach (Table 49-1). The glandular epithelium is divided into cells that secrete products into the gastric lumen for digestion (parietal cells, chief cells, mucus-secreting cells) and cells that control function (gastrin-secreting G cells, somatostatin-secreting D cells) cells. In the cardia, the mucosa is arranged in branched glands and the pits are short. In the fundus and body, the glands are more tubular and the pits are longer. In the antrum, the glands are more branched. The luminal ends of the gastric glands and pits are lined with mucus-secreting surface epithelial cells, which extend down into the necks of the glands for variable distances. In the cardia, the glands are predominantly mucus-secreting. In the body, the glands are mostly lined from the neck to the base with parietal and chief cells (Fig. 49-5). There are a few parietal cells in the fundus and proximal antrum, but none in the cardia or prepyloric antrum. The endocrine G cells are present in greatest quantity in the antral glands.
Table 49-1 Gastric Cell Types, Location, and Function
| CELL TYPE | LOCATION | FUNCTION |
|---|---|---|
| Parietal | Body | Secretion of acid and intrinsic factor |
| Mucus | Body, antrum | Mucus |
| Chief | Body | Pepsin |
| Surface epithelial | Diffuse | Mucus, bicarbonate, prostaglandins (?) |
| Enterochromaffin-like | Body | Histamine |
| G | Antrum | Gastrin |
| D | Body, antrum | Somatostatin |
| Gastric mucosal interneurons | Body, antrum | Gastrin-releasing peptide |
| Enteric neurons | Diffuse | Calcitonin gene–related peptide, others |
| Endocrine | Body | Ghrelin |
Physiology
Gastric Peptides
Gastrin
Gastrin is produced by G cells located in the gastric antrum (see Table 49-1). It is synthesized as a prepropeptide and undergoes post-translational processing to produce biologically reactive gastrin peptides. Several molecular forms of gastrin exist. G-34 (big gastrin), G-17 (little gastrin), and G-14 (minigastrin) have been identified. However, 90% of antral gastrin is released as the 17–amino acid peptide, although G-34 predominates in the circulation because its metabolic half-life is longer than that of G-17. The pentapeptide sequence contained at the carboxyl terminus of gastrin is the biologically active component and is identical to that found on another gut peptide, CCK. CCK and gastrin differ by tyrosine sulfation sites. The release of gastrin is stimulated by food components in a meal, especially protein digestion products. Luminal acid inhibits the release of gastrin. In the antral location, somatostatin and gastrin release are functionally linked, and an inverse reciprocal relationship exists between these two peptides.1
Gastrin is the major hormonal regulator of the gastric phase of acid secretion following a meal. Histamine, released from enterochromaffin-like (ECL) cells, is also a potent stimulant of acid release from the parietal cell. Gastrin also has considerable trophic effects on the parietal cells and gastric ECL cells. Prolonged hypergastrinemia from any cause leads to mucosal hyperplasia and an increase in the number of ECL cells and, under some circumstances, is associated with the development of gastric carcinoid tumors.2
The detection of hypergastrinemia may suggest a pathologic state of acid hypersecretion but generally is the result of treatment with agents to lower acid secretion, such as proton pump inhibitors. Table 49-2 lists common causes of chronic hypergastrinemia. Hypergastrinemia that results from the administration of acid-lowering drugs is an appropriate response caused by loss of feedback inhibition of gastrin release by luminal acid. Lack of acid causes a reduction in somatostatin release, which in turn causes increased release of gastrin from antral G cells. Hypergastrinemia can also occur in the setting of pernicious anemia or uremia, or following surgical procedures such as vagotomy or retained gastric antrum after gastrectomy. In contrast, gastrin levels increase inappropriately in patients with gastrinoma (Zollinger-Ellison syndrome [ZES]). These gastrin-secreting tumors are not located in the antrum and secrete gastrin autonomously.
Table 49-2 Causes of Hypergastrinemia
| ULCEROGENIC CAUSES | NONULCEROGENIC CAUSES |
|---|---|
| Antral G-cell hyperplasia or hyperfunction | Antisecretory agents (PPIs) |
| Retained excluded antrum | Atrophic gastritis |
| Zollinger-Ellison syndrome | Pernicious anemia |
| Gastric outlet obstruction | Acid-reducing procedure (vagotomy) |
| Short-gut syndrome | Helicobacter pylori infection |
| Chronic renal failure |
Somatostatin
Somatostatin is produced by D cells and exists endogenously as the 14– or 28–amino acid peptide. The predominant molecular form in the stomach is somatostatin-14. It is produced by diffuse neuroendocrine cells located in the fundus and antrum. In these locations, D cell cytoplasmic extensions have direct contact with the parietal cells and G cells, where it presumably exerts its actions through paracrine effects on acid secretion and gastrin release.3 Somatostatin is able to inhibit parietal cell acid secretion directly but can also indirectly inhibit acid secretion through inhibition of gastrin release and downregulation of histamine release from ECL cells. The principal stimulus for somatostatin release is antral acidification, whereas acetylcholine from vagal fibers inhibits its release.
Gastric Acid Secretion
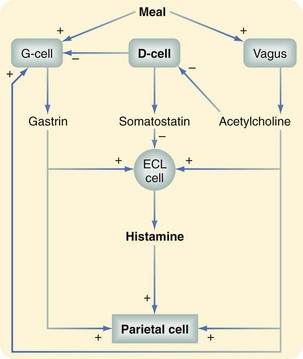
Gastric acid secretion by the parietal cell is regulated by three local stimuli—acetylcholine, gastrin, and histamine. These three stimuli account for basal and stimulated gastric acid secretion. Acetylcholine is the principal neurotransmitter modulating acid secretion and is released from the vagus and parasympathetic ganglion cells. Vagal fibers innervate not only parietal cells but also G cells and ECL cells to modulate release of their peptides. Gastrin has hormonal effects on the parietal cell and stimulates histamine release. Histamine has paracrine-like effects on the parietal cell and, as shown in Figure 49-6, plays a central role in the regulation of acid secretion by the parietal cell after its release from ECL cells. As depicted, somatostatin exerts inhibitory actions on gastric acid secretion. Release of somatostatin from antral D cells is stimulated in the presence of intraluminal acid to a pH of 3 or lower. After its release, somatostatin inhibits gastrin release through paracrine effects and also modifies histamine release from ECL cells. In some patients with PUD, this negative feedback response is defective. Consequently, the precise state of acid secretion by the parietal cell is dependent on the overall influence of the positive and negative stimuli.
Activation and Secretion by the Parietal Cell
The two second messengers principally involved in stimulation of acid secretion by parietal cells are intracellular cyclic AMP (cAMP) and calcium. Synthesis of these two messengers in turn activates protein kinases and phosphorylation cascades. The intracellular events following ligand binding to receptors on the parietal cell are shown in Figure 49-7. Histamine causes an increase in intracellular cAMP, which activates protein kinases to initiate a cascade of phosphorylation events that culminate in activation of H+, K+-ATPase. In contrast, acetylcholine and gastrin stimulate phospholipase C, which converts membrane-bound phospholipids into inositol triphosphate (IP3) to mobilize calcium from intracellular stores. Increased intracellular calcium activates other protein kinases that ultimately activate H+, K+-ATPase in a similar fashion to initiate the secretion of hydrochloric acid.
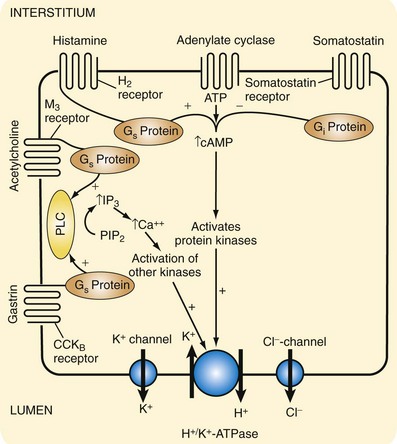
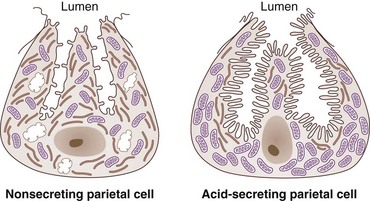
H+, K+-ATPase is the final common pathway for gastric acid secretion by the parietal cell. It is composed of two subunits, a catalytic α-subunit (100 kDa) and a glycoprotein β-subunit (60 kDa). During the resting, or nonsecreting, state, gastric parietal cells store H+, K+-ATPase within intracellular tubulovesicular elements. Cellular relocation of the proton pump subunits through cytoskeletal rearrangements must occur in order for acid secretion to increase in response to stimulatory factors. The subsequent insertion and heterodimer assembly of the H+, K+-ATPase subunits into the microvilli of the secretory canaliculus causes an increase in gastric acid secretion. A KCl efflux pathway must exist to supply potassium to the extracytoplasmic side of the pump. Cytosolic hydrogen is secreted by H+, K+-ATPase in exchange for extracytoplasmic potassium (see Fig. 49-7), which is an electroneutral exchange and therefore does not contribute to the transmembrane potential difference across the parietal cell. Secretion of chloride is accomplished through a chloride channel moving chloride from the parietal cell cytoplasm to the gastric lumen. The secretion or exchange of hydrogen for potassium, however, does require energy in the form of adenosine triphosphate (ATP) because hydrogen is being secreted against a gradient of more than a million-fold. Because of this large energy requirement, the parietal cell also has the largest mitochondrial content of any mammalian cell, with a mitochondrial compartment representing 34% of its cell volume. In response to a secretagogue, the parietal cell undergoes a conformational change, and a several-fold increase in the canalicular surface area occurs (Fig. 49-8). In contrast to stimulated acid secretion, cessation of acid secretion requires endocytosis of H+, K+-ATPase, with regeneration of cytoplasmic tubulovesicles containing the subunits, and this occurs through a tyrosine-based signal. The tyrosine-containing sequence is located on the cytoplasmic tail of the β-subunit and is highly homologous to the motif responsible for internalization of the transferrin receptor.
Pharmacologic Regulation
One notable side effect of all antisecretory agents is the elevation of serum gastrin levels. Serum gastrin levels are higher after treatment with PPIs than with H2 receptor antagonists. This effect is accompanied by hyperplasia of G cells and ECL cells when these agents are administered chronically. Chronic administration of omeprazole has been found to cause ECL hyperplasia that could progress to carcinoid tumors in rats.3 This effect, however, was not specific for omeprazole and was reproduced by other agents that caused prolonged inhibition of acid secretion and resultant hypergastrinemia.
Gastric Motility
Gastric motility is regulated by extrinsic and intrinsic neural mechanisms and by myogenic control. The extrinsic neural controls are mediated through parasympathetic (vagus) and sympathetic pathways, whereas the intrinsic controls involve the enteric nervous system (see earlier, “Anatomy”). In contrast, myogenic control resides in the excitatory membranes of the gastric smooth muscle cells.
Abnormal Gastric Motility
Symptoms of abnormal gastric motility are nausea, fullness, early satiety, abdominal pain, and discomfort. Although mechanical obstruction can and should be ruled out with upper endoscopy or radiographic contrast studies, objective evaluation of a patient with a suspected motility disorder can be accomplished with gamma scintigraphy, real-time ultrasound, and magnetic resonance imaging (MRI). Gastric motility disorders usually encountered in clinical practice are gastric dysmotility following vagotomy, delayed gastric emptying associated with diabetes mellitus, and gastric motility dysfunction related to H. pylori infection. Vagotomy results in loss of receptive relaxation and gastric accommodation in response to meal ingestion, with resultant early satiety, postprandial bloating, accelerated emptying of liquids, and delay in emptying of solids. Clinical manifestations of diabetic gastropathy, which can occur in insulin-dependent or insulin-independent patients, closely resemble the clinical picture of postvagotomy gastroparesis. Furthermore, structural changes have been identified in the vagus nerve of patients with diabetes, suggesting that a diabetic autonomic neuropathy may be responsible. However, the metabolic effects of diabetes have also been implicated. Specifically, hyperglycemia has been shown to cause a decrease in contractility of the gastric antrum, increase in pyloric contractility, and suppression of the migrating motor complex (MMC). Suppression of MMC activity is thought to be responsible for the accumulation of gastric bezoars seen in some diabetic patients. In contrast, hyperinsulinemia, which is often associated with non–insulin-dependent diabetes, may play a role in the gastroparesis seen in non–insulin-dependent diabetes because it also leads to suppression of MMC activity.4
H. pylori–infected patients with nonulcer dyspepsia have also been demonstrated to have impaired gastric emptying accompanied by a reduction in gastric compliance.5 In rats, lipopolysaccharide derived from H. pylori causes a reduction in gastric emptying of a liquid meal for up to 12 hours by an unknown mechanism. Regardless of the cause of gastroparesis, treatment consists of prokinetic agents, such as metoclopramide and erythromycin, which have been shown to have some benefit, although the evidence is more compelling in diabetics.
Peptic Ulcer Disease
Epidemiology
The estimated prevalence of PUD ranges from 5% to 15% in Western populations, with a lifetime incidence of almost 10%.6 Although the incidence and hospitalization rate for PUD have been decreasing since the 1980s, it remains one of the most prevalent and costly GI diseases. Medical costs associated with PUD are an estimated $5.65 billion annually. An estimated 15,000 operations are performed each year on patients hospitalized with PUD. Significant progress has been made over the past 2 decades, with total admissions for PUD decreasing by almost 30%. Admissions for complications of ulcer disease have also been decreasing, which has led to a significant decrease in ulcer-related mortality, from 3.9% in 1993 to 2.7% in 2006.7 Although overall mortality remains low, this still represents over 4000 deaths caused by PUD each year.
The role of surgery in the treatment of ulcer disease has also decreased, primarily caused by a marked decline in elective surgical therapy for chronic disease because the percentage of patients who require emergent surgery for complicated disease has remained constant, at 7% of hospitalized patients.7 This represents over 11,000 surgical procedures annually.
Pathogenesis
Peptic ulcers are caused by increased aggressive factors, decreased defensive factors, or both.8 This in turn leads to mucosal damage and subsequent ulceration. Protective (or defensive) factors include mucosal bicarbonate secretion, mucus production, blood flow, growth factors, cell renewal, and endogenous prostaglandins. Damaging (or aggressive) factors include hydrochloric acid secretion, pepsins, ethanol ingestion, smoking, duodenal reflux of bile, ischemia, NSAIDs, hypoxia and, most notably, H. pylori infection.
Helicobacter pylori Infection
Duodenal Ulcer
Diagnosis
Upper Gastrointestinal Radiography
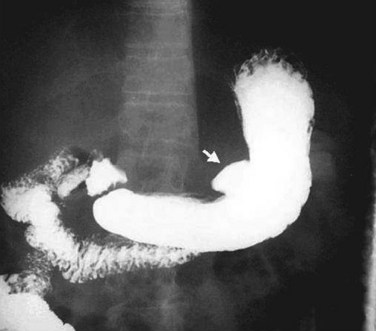
Diagnosis of duodenal ulcer by upper GI radiography requires the demonstration of barium within the ulcer crater, which is usually round or oval and may or may not be surrounded by edema. This study is useful to determine the location and depth of penetration of the ulcer and the extent of deformation from chronic fibrosis. A characteristic barium radiograph of a peptic ulcer is shown in Figure 49-9. The ability to detect ulcers on radiography requires the technical skills and abilities of the radiologist but is also dependent on the size and location of the ulcer. With single-contrast radiographic techniques, as many as 50% of duodenal ulcers may be missed, whereas with double-contrast studies, 80% to 90% of ulcer craters can be detected.
Treatment
Medical Management
Treatment of Helicobacter pylori Infection
Current therapy is twofold in its approach, combining antibiotics against H. pylori with antacid medications. The primary goal of the antacids is to promote short-term healing by reducing pathologic acid levels and improve symptoms. H. pylori eradication helps with initial healing, but its primary efficacy is in preventing recurrence. There have been numerous trials comparing eradication therapy with ulcer-healing drugs alone or no treatment. Eradication of H. pylori has shown recurrence rates as low as 2%, with initial healing as high as 90%. This compares with recurrence rates of up to 25% with ulcer-healing medications alone. One review has analyzed the results of these trials and further validated the role of antibiotics in the treatment of H. pylori–positive duodenal ulcers.10 Both eradication therapy and ulcer-healing drugs alone have shown initial healing rates higher than 80%. Eradication therapy has resulted in long-term recurrence of less than 15%, in contrast to 64% recurrence in patients treated with only a short initial course of ulcer-healing drugs. Patients could achieve low recurrence rates similar to those of eradication therapy, but only if they were maintained on their antacid regiment long term, as opposed to a 1- or 2-week course of eradication therapy.
Given all these findings across multiple studies, and according to the recommendations of the American Gastroenterological Association, European Helicobacter pylori Study Group, and National Institutes of Health (NIH), the treatment of H. pylori–positive peptic duodenal ulcer disease is triple therapy aimed at the eradication of H. pylori, along with acid suppression (Box 49-1). This includes an antisecretory agent, now most commonly a PPI, although histamine antagonists are still used, along with two antibiotics, usually amoxicillin with clarithromycin or metronidazole, given for a 2-week course. Side effects, which are generally mild and resolve with cessation of treatment, include diarrhea, nausea and vomiting, rash, and altered taste. For the 10% of patients with refractory disease, quadruple therapy with the addition of bismuth is recommended.
Complicated Ulcer Disease
Ulcer disease was once very much the purview of the general surgeon, with ulcer surgery forming a major part of general surgery practice. With the shift in understanding of the disease from one primarily of aberrant acid physiology to one of infectious disease, this has changed significantly, with the overwhelming majority of ulcer patients being treated and cured medically. The surgeon’s role now is primarily to treat the approximately 20% of patients who have a complication from their disease, which includes hemorrhage, perforation, and obstruction (Box 49-2). Frequently included in discussions of complicated ulcer disease is the intractable ulcer. Although intractable disease no doubt exists, its definition is nebulous and exactly when and what type of surgical intervention it requires remain primarily a matter of judgment.
Box 49-2 Surgical Treatment Recommendations for Complications Related to Peptic Duodenal Ulcer Disease
Intractable: Parietal cell vagotomy ± antrectomy
Bleeding: Oversewing of bleeding vessel with treatment of H. pylori
Perforation: Patch closure with treatment of H. pylori
Obstruction: Rule out malignancy and gastrojejunostomy with treatment of H. pylori
Hemorrhage
Upper GI bleeding remains a relatively common problem, with an annual incidence of approximately 1/1000.11 Most nonvariceal bleeding (70%) is attributable to peptic ulcers. Most bleeding will stop spontaneously and requires no intervention; persistent bleeding, however, is associated with a 6% to 8% mortality.
The primary clinical criteria that predict persistent bleeding or rebleeding after initial cessation of bleeding, and thus increased mortality, are increased age, decreased hemoglobin (<10 g/dL) at presentation, shock, melena, and need for blood transfusion. Patients who meet any of these criteria should be considered as high risk.11
Patients who are noted to have active bleeding, via an arterial jet or oozing, an adherent clot, or a visible vessel within the ulcer, are at high risk and intervention is required. Patients without active bleeding, no visible vessel, and a clean ulcer base are low risk and do not require further intervention. All patients undergoing endoscopic examination should be tested for their H. pylori status. For the high-risk patient requiring intervention, the best initial approach is endoscopic control, which results in primary hemostasis in approximately 90% of patients. The most common method of control is injection of a vasoconstrictor at the site of bleeding. However, with this method alone, primary hemostasis rates are high but up to 30% of patients have rebleeding. This has led to the development of new techniques, including use of a second vasoconstrictor or sclerosing agent, thermal coagulation, and placement of clips at the site of bleeding. A 2007 meta-analysis has compared the use of epinephrine alone to epinephrine plus any second technique.12 A dual approach, when compared with epinephrine alone, showed better primary hemostasis, reduction in rebleeding rate, lower rate of surgery, and decreased mortality.
Importantly, both thermal coagulation and mechanical clips, in single-method comparisons with epinephrine, have shown significant superiority with respect to primary hemostasis and rebleeding. A meta-analysis comparing either of these two techniques alone to the use of dual methods has shown no significant difference, except in cases of active arterial bleeding, in which case use of a second method was superior. Although the cost and complications from epinephrine remain small, the use of dual methods does have slightly higher, although still less than 1%, complication rates (e.g., necrosis, perforation), than any single technique. Current 2003 guidelines for endoscopic control of bleeding advocate the use of epinephrine plus an additional method.11 As more data become available, it may be demonstrated that thermal or mechanical treatment alone can be used for most patients. For patients who have rebleeding, repeat endoscopy does not increase their mortality and should be attempted prior to surgical intervention.
All high-risk patients should be placed in a monitored setting, preferably an intensive care unit, until all bleeding has stopped for 24 hours. As part of the 2003 consensus guidelines, all high-risk patients should be placed on an IV PPI, with an initial bolus followed by continuous infusion or intermittent dosing for up to 72 hours. When compared with a histamine blocker and placebo, the IV PPI showed lower rebleeding rates, lower rate of emergency surgery, and decreased mortality.13 Patients deemed high risk based on clinical factors who are awaiting endoscopy should probably begin therapy, even prior to endoscopy.
Despite the use of PPIs and improved methods of endoscopic control, 5% to 10% of patients will have persistent bleeding that requires surgical intervention. The vessel most likely to be bleeding is the gastroduodenal artery because of erosion from a posterior ulcer. The duodenum is opened longitudinally, with the incision carried across the pylorus. The vessel is oversewn, with a three-point U stitch technique, which effectively ligates the main vessel along with any smaller branches. One must be careful to avoid incorporating the common bile duct into the stitch. The duodenotomy is closed transversely to avoid narrowing (Fig. 49-10).
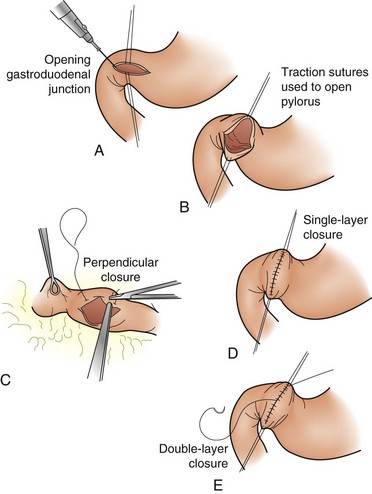
FIGURE 49-10 A-E, Heineke-Mikulicz pyloroplasty.
(From Soreide JA, Soreide A: Pyloroplasty. Oper Tech Gen Surg 5:65–72, 2003.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


