End-stage hypertrophic cardiomyopathy (ES-HC) has an ominous prognosis. Whether genotype can influence ES-HC occurrence is unresolved. We assessed the spectrum and clinical correlates of HC-associated mutations in a large multicenter cohort with end-stage ES-HC. Sequencing analysis of 8 sarcomere genes (MYH7, MYBPC3, TNNI3, TNNT2, TPM1, MYL2, MYL3, and ACTC1) and 2 metabolic genes (PRKAG2 and LAMP2) was performed in 156 ES-HC patients with left ventricular (LV) ejection fraction (EF) <50%. A comparison among mutated and negative ES-HC patients and a reference cohort of 181 HC patients with preserved LVEF was performed. Overall, 131 mutations (36 novel) were identified in 104 ES-HC patients (67%) predominantly affecting MYH7 and MYBPC3 (80%). Complex genotypes with double or triple mutations were present in 13% compared with 5% of the reference cohort (p = 0.013). The distribution of mutations was otherwise indistinguishable in the 2 groups. Among ES-HC patients, those presenting at first evaluation before the age of 20 had a 30% prevalence of complex genotypes compared with 19% and 21% in the subgroups aged 20 to 59 and ≥60 years (p = 0.003). MYBPC3 mutation carriers with ES-HC were older than patients with MYH7, other single mutations, or multiple mutations (median 41 vs 16, 26, and 28 years, p ≤0.001). Outcome of ES-HC patients was severe irrespective of genotype. In conclusion, the ES phase of HC is associated with a variable genetic substrate, not distinguishable from that of patients with HC and preserved EF, except for a higher frequency of complex genotypes with double or triple mutations of sarcomere genes.
The end-stage (ES) phase of hypertrophic cardiomyopathy (HC) carries an ominous prognosis because of high rates of refractory heart failure and sudden arrhythmic death and represents the sole indication for heart transplantation in HC. Several different mechanisms have been proposed to explain the evolution toward ES, such as progressive cardiomyocyte energy depletion, microvascular ischemia, and replacement fibrosis although it remains largely unresolved. It has also been hypothesized that development of ES may be heavily influenced by the specific genetic background. Indeed, multiple sarcomere gene mutations are associated with early onset of HC disease and particularly severe phenotypes, including evolution toward the ES. To date, however, systematic studies on the genetic background of ES-HC are limited, probably because of the relative rarity of this condition in most HC cohorts. Therefore, to adequately investigate this issue, we assessed the prevalence and spectrum of sarcomere gene mutations in the largest multicenter cohort of patients with ES-HC reported to date, with respect to phenotypic expression, clinical course, and outcome. Moreover, a comparison of the genetic substrate in ES-HC with a subset of HC patients with preserved left ventricular (LV) ejection fraction (EF) was assessed.
Methods
In this multicenter cross-sectional and longitudinal study, we retrospectively identified 156 patients diagnosed with ES-HC (130 index cases and 26 family members) from January 1981 to June 2010 and genetically screened between January 2007 and June 2010 at 6 referral centers in Italy and 2 in the United States. Five of these centers (Dipartimento di Medicina Specialistica, Diagnostica e Sperimentale, Alma Mater Studiorum, Università di Bologna, Italy; Dipartimento di Cardiologia, Seconda Università degli Studi, Napoli, Italy; Dipartimento Cardiovascolare, Ospedali Riuniti, Bergamo, Italy; Hypertrophic Cardiomyopathy Center, Minneapolis Heart Institute Foundation, Minneapolis, MN; Tufts Medical Center, Hypertrophic Cardiomyopathy Center, Boston, MA) have dedicated heart failure management units including heart transplantation programs.
Diagnosis of HC was based on echocardiographic and/or cardiovascular magnetic resonance imaging documentation of a hypertrophied, nondilated LV, in the absence of other cardiac or systemic diseases that could produce the magnitude of LV hypertrophy evident. ES-HC was defined as 2D echocardiographic LV EF <50% at rest, reflecting global systolic dysfunction, at study entry or during follow-up. Patients with previous surgical myectomy or alcohol septal ablation (n = 16), known atherosclerotic coronary artery disease (n = 9), or severe valvular heart disease (n = 8) were excluded.
A subset of 181 patients with HC and normal LV systolic function consecutively genotyped for 10 genes between January 2007 and June 2010 at Centro di Riferimento per le Cardiomiopatie, Azienda Ospedaliero-Universitaria Careggi, Firenze, Italy, was examined as a reference cohort. Median age at first evaluation was 52 years (interquartile range [IQR] 36 to 63); 64 (35%) were female, median maximum LV wall thickness was 22 mm (IQR 19 to 27), left atrium diameter 45 mm (IQR 40 to 51); 34 (19%) had LV outflow tract obstruction in conditions at rest; 164 of 181 patients (91%) were in New York Heart Association (NYHA) class I to II; 25 (19%) were implanted with an implantable cardioverter-defibrillator (ICD) for primary or secondary protection from sudden death.
We considered echocardiographic studies performed at first evaluation at the referral center and first documentation of ES. Maximum wall thickness, LV end-diastolic cavity dimension, left atrial dimension, LV outflow tract obstruction, and LVEF were evaluated. LV hypertrophy was assessed with 2-dimensional echocardiography, and the site of maximum wall thickness was identified.
All patients underwent genetic testing for the 8 most frequently mutated sarcomeric genes associated with HC: cardiac beta-myosin heavy chain (MYH7, NM_000257.2), cardiac myosin-binding protein C (MYBPC3, NM_000256.3), troponin I (TNNI3, NM_000363.4), troponin T (TNNT2, NM_001001430.1), alpha-tropomyosin (TPM1, NM_000366.5), regulatory myosin light chain (MYL2, NM_000432.3), essential myosin light chain (MYL3, NM_000258.2), and alpha-cardiac actin (ACTC1, NM_005159.4) as well for lysosome-associated membrane protein 2 (LAMP2, NM_002294.2) and AMP-activated protein kinase (PRKAG2, NM_016203.3) as possible cause of HC phenocopies. Written informed consent was obtained in each case. Conventional DNA sequencing was performed using standard method.
Each identified variant was confirmed by direct sequencing from an independent amplification product and, whenever possible, by restriction enzyme digestion. The significance of each variation was defined by the following criteria, although not always all of them present for each mutation: (1) absence in 300 adult control chromosomes from ethnically matched subjects, tested by sequencing; (2) minor allele frequency (MAF) <1%, collected from ESP (Exome Sequencing Project, http://evs.gs.washington.edu/EVS/ , data release ESP6500SI-V2) and dbSNP137 ( http://www.ncbi.nlm.nih.gov/SNP/ ), that includes 1000Genomes Project data; (3) evolutionary conservation of the nucleotide or amino acid, calculated by multiple alignments of 46 vertebrate species and measured by phyloP score; and (4) in silico functional prediction of effect by in silico tools: PolyPhen ( http://genetics.bwh.harvard.edu/pph/ ), SIFT ( http://sift.jcvi.org/ ), and MutationTaster ( http://neurocore.charite.de/MutationTaster ) for coding variants; a specific software Alamut 2.0 was used to search for splicing prediction. Moreover, the presence in the literature of identified variant was recorded. When family members were available, cosegregation of the mutation with disease was tested, especially for complex genotype.
Cardiovascular mortality was defined as death because of cardiovascular causes, including sudden cardiac death, heart failure–related death, and stroke-related death. Deaths because of cardiac complications after heart transplantation (such as acute rejection) were also included.
HC-related death or equivalents: cumulative end point consisting of cardiovascular mortality (as previously defined) and cardiac death equivalents including appropriate ICD intervention for ventricular fibrillation or ventricular tachycardia with a heart rate ≥200 beats/min and cardiac transplantation.
Continuous variables were expressed as median and IQR and were compared with Mann-Whitney rank-sum test or Kruskal-Wallis test. Noncontinuous variables were expressed as proportions and were compared with Pearson’s chi-square test. Probability values were considered significant when ≤0.05. Univariate and multivariate logistic regression analysis were used to identify baseline clinical predictors of cardiovascular mortality and HC-related death or equivalents. Probability of death-free survival was calculated using the Kaplan-Meier method, and survival curves were compared using the log-rank test.
Results
A total of 156 patients was identified with ES-HC ( Table 1 ). Ninety-two patients (59%) had only mild or no symptoms (NYHA functional class I to II) at the time of ES diagnosis. Sequencing analysis identified a total of 131 mutations in 104 ES-HC patients (overall prevalence 67%) including 101 missense, 13 splicing, 11 frameshift, 4 nonsense, and 2 in-frame deletion mutations. Of these, 95 (73%) mutations have been previously reported as associated with HC phenotype, whereas 36 (27%) were novel variants. The majority occurred in MYBPC3 (n = 71, 5%), MYH7 (n = 34, 26%), and TNNI3 (n = 12, 9%), whereas mutations in TPM1, TNNT2, ACTC1, MYL3, MYL2, and LAMP2 were uncommon and collectively accounted for 11% of all identified variants. Eighty-three patients (53%) had single mutations (38 in MYBPC3, 26 in MYH7, 9 in TNNI3, 3 in TPM1, 3 in TNNT2, 2 in MYL2, and 1 in MYL3 and LAMP2 each), whereas 21 patients (13%) had a complex genotype characterized by double-gene mutation heterozygosis (n = 6; 4%), compound heterozygosis (n = 9; 6%), triple mutations (n = 4; 3%), and homozygosis (n = 2; 1%) ( Figure 1 ). The gene most frequently involved in complex genotype was MYBPC3 (19 of 21 complex genotypes, 90%). A novel LAMP2 mutation probably altering the splicing was identified in 1 patient. No mutations were identified in PRKAG2.
| Variable | Overall | Negative Genotype | Single Mutations | Multiple Mutations | p Value |
|---|---|---|---|---|---|
| (n = 156) | (n = 52) | (n = 83) | (n = 21) | ||
| Proband | 130 (83%) | 50 (96%) | 63 (76%) | 17 (81%) | 0.008 |
| Male sex | 86 (55%) | 29 (56%) | 43 (52%) | 14 (67%) | 0.470 |
| Age at HC diagnosis (years) | 30 (16–45) | 34 (17–50) | 30 (16–47) | 28 (17–33) | 0.271 |
| Age at 1st evaluation (years) | 44 (30–55) | 45 (32–55) | 45 (31–55) | 40 (26–46) | 0.317 |
| Age at ES diagnosis (years) | 45 (35–57) | 47 (33–58) | 47 (36–58) | 42 (35–51) | 0.342 |
| Family history of HC | 97 (63%) | 26 (50%) | 59 (71%) | 12 (60%) | 0.047 |
| Family history of SD | 57 (37%) | 16 (31%) | 35 (42%) | 6 (30%) | 0.326 |
| Family history of ES HC | 45 (29%) | 11 (21%) | 27 (33%) | 7 (35%) | 0.300 |
| Echocardiographic data at ES diagnosis | |||||
| Maximal wall thickness (mm) | 17 (14–22) | 17 (14–22) | 17 (14–20) | 17 (15–19) | 0.702 |
| LV ED cavity dimension (mm) | 55 (49–59) | 55 (49–59) | 50 (47–55) | 55 (51–61) | 0.019 |
| LVEF (%) | 45 (34–48) | 45 (34–48) | 45 (36–49) | 42 (33–45) | 0.425 |

The MAF value was collected from ESP and dbSNP137, which also includes the 1000Genome Project data. Based on this analysis, no mutation (novel or previously reported) had an MAF >1%. In particular, none of our novel mutations is present in any publicly available database (MAF = 0), whereas among the previously reported mutations, 16 have been reported as very rare, showing an MAF between 0.0077% and 0.0392% but depicted as probably or possibly damaging. Furthermore, we had found also c.83 C > T and c.833 G > A in TNNT2 and MYBPC3 gene, respectively, previously classified as mutations. These 2 variants are now reported in ESP database as benign; therefore, we decided to eliminate them.
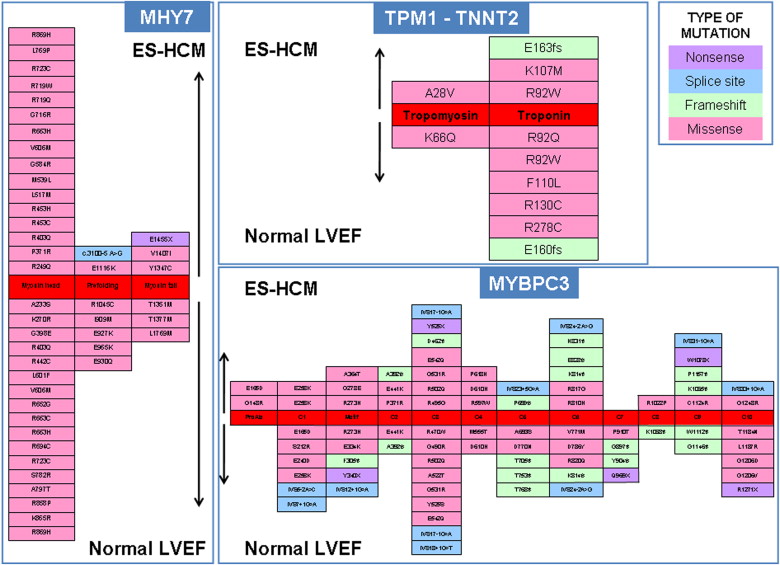
Compared with the 181 patients with HC phenotype and preserved LVEF, patients with ES-HC showed a greater prevalence of complex genotypes (21/156 or 13% vs 9/181 or 5%, p = 0.013); otherwise, the overall prevalence and gene distribution of mutations were not different in the 2 groups; specifically, the location of mutations and the mutated residues were comparable ( Figure 2 ). Clinical and echocardiographic features did not differ significantly between genotype-positive and genotype-negative ES-HC patients, except for prevalence of HC family history ( Table 1 ). Although median age at first evaluation was 5 years younger, patients with complex genotypes were comparable with those with single or no mutations regarding clinical or demographic features. Regarding the specific genes involved, patients with single mutations in MYBPC3 gene were older at HC diagnosis, first evaluation, and ES diagnosis compared with those with single mutations in MYH7, single mutations in other genes, or with multiple mutations ( Table 2 ). Patients with single mutations in genes different from MYBPC3 or MYH7 (mostly TNNI3) showed a higher LVEF at first detection of ES compared with patients with single mutations in MYBPC3, MYH7, or complex genotypes (47% [IQR 43% to 49%] vs 45% [IQR 35% to 49%], 40% [IQR 35% to 45%], and 42% [IQR 33% to 45%], respectively, p = 0.018) ( Table 2 ). Of note, there was a 30% prevalence of patients with complex genotypes among those presenting at first evaluation before the age of 20 compared with 19% and 21% in the subgroups aged 20 to 59 years and ≥60 years, respectively (p = 0.003). Conversely, single MYBPC3 mutations were by far the most prevalent genotype among patients ≥60 years at first evaluation (79% vs 20% and 31% in the other age subgroups, p = 0.003).
| Variable | Single Mutations in MYBPC3 | Single Mutations in MYH7 | Other Single Mutations | Multiple Mutations | p Value |
|---|---|---|---|---|---|
| (n = 38) | (n = 26) | (n = 19) | (n = 21) | ||
| Proband | 32 (84%) | 16 (61%) | 15 (79%) | 17 (81%) | 0.184 |
| Male sex | 22 (58%) | 11 (42%) | 10 (53%) | 14 (67%) | 0.389 |
| Age at HC diagnosis (years) | 41 (25–55) | 16 (10–30) | 26 (11–44) | 28 (17–33) | 0.0001 |
| Age at 1st evaluation (years) | 53 (36–62) | 40 (29–45) | 39 (20–51) | 40 (26–46) | 0.002 |
| Age at ES diagnosis (years) | 54 (45–64) | 41 (30–48) | 38 (29–56) | 42 (35–51) | 0.001 |
| Family history of HC | 25 (66%) | 20 (77%) | 14 (74%) | 12 (57%) | 0.594 |
| Family history of SD | 11 (29%) | 14 (54%) | 10 (53%) | 6 (30%) | 0.106 |
| Family history of ES HC | 8 (21%) | 13 (50%) | 6 (32%) | 7 (35%) | 0.116 |
| NYHA III-IV | 24 (63%) | 19 (73%) | 12 (63%) | 13 (62%) | 0.821 |
| Echocardiographic data at ES diagnosis | |||||
| Maximal wall thickness (mm) | 17 (14–21) | 18 (14–20) | 15 (13–18) | 17 (15–19) | 0.103 |
| LV ED cavity dimension (mm) | 52 (47–57) | 50 (48–53) | 49 (44–56) | 54 (51–61) | 0.087 |
| LVEF (%) | 40 (35–45) | 45 (35–49) | 47 (43–49) | 42 (33–45) | 0.018 |
In 95 of 104 patients with positive genotype, a time course from the diagnosis of HC with preserved systolic function to ES evolution was identifiable. In these 95 patients, median LVEF at first evaluation was 60% (IQR 58 to 66), maximum wall thickness 22 mm (IQR 18 to 26), LV end-diastolic diameter was 45 mm (IQR 40 to 50), LV outflow obstruction was present in 14 (15%), and massive LV hypertrophy (maximum wall thickness ≥30 mm) in 10 (10%). Progression to ES was characterized by a 25% reduction in EF (from a median value of 60% to 45%), a 13% increase in LV end-diastolic diameter (from a median value of 45 to 51 mm), and a 23% reduction in LV thickness (from a median value of 22 to 17 mm) during a median interval of 16 years.
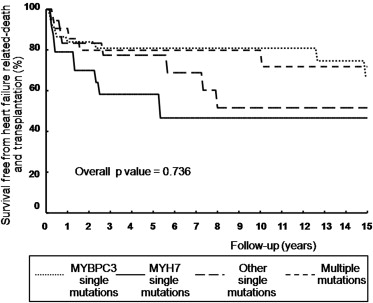
Among the 95 ES-HC patients with normal systolic function at initial presentation, median age at presentation was 16 years (IQR 10 to 32) for patients with single mutations in MYH7 gene, compared with 26 years (IQR 11 to 45) in patients with single mutations in other genes including other thick filament genes. Conversely, patients with single MYBPC3 mutations were older both at HC diagnosis and at ES diagnosis compared with patients with single MHY7 mutations, other single-gene mutations, and multiple mutations (p = 0.0002 and p = 0.0008, respectively; Figure 3 ).

Median duration of follow-up from first detection of ES was 3.6 years (IQR 1.18 to 8.02). During follow-up, 103 patients (66%) progressed to NHYA functional class III to IV. Overall, 26 patients died (17%): 4 experienced sudden death, 14 because of heart failure, 2 because of stroke, 2 because of noncardiac causes, and 4 after cardiac transplantation. Of note, 7 of these 26 patients had had 1 or more appropriate ICD interventions before death. Therefore, 130 patients (83%) were alive at the end of follow-up. In comparison, 73 patients (47%) reached the end point HC-related death or equivalents. In addition to the 25 patients who died, 44 underwent heart transplantation (including 3 with previous ICD interventions), and 4 patients (who were alive at the end of follow-up) experienced appropriate ICD discharges. The total number of patients who experienced appropriate ICD interventions during follow-up was 14 (9% or 2.5% per year). In the light of these findings, only 53% of ES-HC patients would have plausibly been alive at the end of follow-up in the absence of aggressive management. Of note, cardiac transplantation was associated with favorable outcome in 44 of 48 cases (92%), who were alive after a median of 7.4 years (IQR 4.3 to 12.1). Of the 4 patients who died in this subset, 1 died of acute rejection and 3 of noncardiac causes, respectively, at 1 month, 6 years, and 18 years after transplantation.
Outcome was similar both in terms of cardiovascular death or HC-related death in genotype-positive and genotype-negative patients (16% vs 8%, p = 0.213, and 46% vs 48%, p = 0.253, respectively). Similarly, no significant differences were present between patients with single mutations in MYH7, MYBPC3, other single gene mutations, or complex genotypes (p = 0.65). At multivariate analysis, age at initial evaluation (hazard ratio [HR] 1.03; confidence interval [CI] 1.01 to 1.07, p = 0.03) and NYHA III to IV at initial evaluation (HR 3.04, CI 1.23 to 7.51, p = 0.02) were independently associated with cardiovascular death. Furthermore, age at ES diagnosis, family history of ES-HC, LVEF at initial evaluation, and NYHA III to IV at initial evaluation were independently associated with HC-related death or equivalents (HR 0.97, CI 0.95 to 0.99, p = 0.002; HR 1.79, CI 1.03 to 3.10, p = 0.036; HR 0.97, CI 0.95 to 0.99, p = 0.018; and HR 2.53, CI 1.45 to 4.41, p = 0.001, respectively). Kaplan-Meier estimates of the proportion of patients without heart failure–related death and cardiac transplantation according to the structural characteristics of mutated proteins are shown in Figure 4 .