Signal Transduction: Proliferative Signaling
The signaling systems that regulate cell size, shape, and composition play an important role in the pathophysiology of cardiac disease. These systems, along with those that control cell division, are described as proliferative in this text to distinguish them from the functional systems that modify interactions among preexisting structures. Functional and proliferative signaling was once thought to be controlled by different, independently regulated mechanisms. However, as noted by Bourne (1995), instead of “a few discrete clans” of signaling molecules organized in distinct functional and proliferative pathways, control of these two types of response resembles a series of “wheels within wheels! … bustling communication networks within and between clans of signaling proteins ….” For example, the G protein-coupled receptors initially identified as functional regulators (see Chapter 8) are now known to modify proliferative responses once thought to be under the exclusive control of receptor tyrosine kinases. Conversely, the receptor tyrosine kinases initially viewed as mediating only proliferative responses have been found to influence such functional variables as myocardial contractility and vascular tone. The clinical importance of this crosstalk is enormous. It helps explain why β-adrenergic blockers, whose negative inotropic effects have obvious short-term deleterious effects in systolic heart failure, improve long-term outcome (see Chapter 18), and why direct-acting vasodilators, even though they reduce afterload, often worsen prognosis in these patients. In accord with the principle that what is obvious is not always important, and what is important is not always obvious, formerly overlooked proliferative effects of the neurohumoral response (see Chapter 8) can be of greater clinical significance than the more obvious functional effects.
Can Cardiac Myocytes Divide?
Unlike the connective tissue cells of adult mammalian hearts, which undergo rapid proliferation, cardiac myocytes rarely divide, and synthesis of DNA and protein is slow—the half-lives of myofibrillar proteins, for example, can be several days. However, these myocytes have not entirely lost their ability to proliferate. In a classical review, Rumyantsev (1977) lists histological studies going back to the 1880s which show that when severely stressed, cardiac myocytes have a limited capacity for mitosis (Fig. 9-1). These findings have been confirmed in recent studies using the tools of modern molecular biology (Nadal-Ginard et al., 2003). It remains clear, however, that this limited capacity for cell division cannot regenerate significant amounts of functioning myocardium.
Although adult mammalian hearts cannot adapt to sustained overload or myocyte death by increasing myocyte number (hyperplasia), cardiac myocytes readily enlarge (hypertrophy). This hypertrophic response is well suited for the heart, which cannot suspend its pumping to
incorporate newly generated myocytes into the intricate architecture of its walls (see Chapter 1). As noted by Goss (1966): “By giving up the potential for hyperplasia in favor of the necessity for constant function, [the heart … has] adapted a strategy that enables [it] to become hypertrophic to a limited extent while doing [its jobs] efficiently.” However, a “price” must be paid; because cardiac myocytes have virtually no ability to divide, patients with damaged or overloaded hearts must depend on myocyte hypertrophy to develop an adequate wall stress. Unfortunately, the proliferative responses that allow the adult heart to hypertrophy both damage these cells and shorten cardiac myocyte survival (see Chapter 18).
incorporate newly generated myocytes into the intricate architecture of its walls (see Chapter 1). As noted by Goss (1966): “By giving up the potential for hyperplasia in favor of the necessity for constant function, [the heart … has] adapted a strategy that enables [it] to become hypertrophic to a limited extent while doing [its jobs] efficiently.” However, a “price” must be paid; because cardiac myocytes have virtually no ability to divide, patients with damaged or overloaded hearts must depend on myocyte hypertrophy to develop an adequate wall stress. Unfortunately, the proliferative responses that allow the adult heart to hypertrophy both damage these cells and shorten cardiac myocyte survival (see Chapter 18).
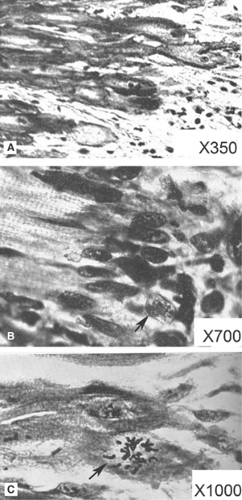 Fig. 9-1: Histology at the edge of experimental myocardial infarctions produced by coronary artery ligation in rabbit hearts. A: 5 days after ligation showing the infarcted area (right) and surviving myocytes whose ends are slightly expanded (left). B: 16 days after ligation showing fibroblast nuclei surrounding the ends of the surviving myocytes, one of whose nuclei is vesicular (arrow). C: 16 days after ligation showing a surviving myocyte undergoing mitosis (arrow). (Modified from Ring, 1950). |
The Cell Cycle
Cells lose their ability to divide when they withdraw from the cell cycle. The latter is a highly regulated process in which cell enlargement, DNA replication, and nuclear division (karyokinesis) are followed by cell division (cytokinesis) that produces two daughter cells, each of which contains a set of genes copied from the mother cell. The cell cycle plays a critical role in the survival of simple life forms, called prokaryotes (the name comes from the absence of a formed nucleus) that lack the internal membrane structures needed to control their milieu intárieur. Instead, prokaryotes can survive environmental change because their ability to divide rapidly allows them grow their way out of trouble. Because each prokaryote represents an independent entity with its own complement of genes, when a population of these organisms encounters a major environmental change, selection of cells whose phenotype is best able to withstand the new stress helps some of the population to survive. Even if a vast majority dies, rapid cell cycling allows a few survivors to multiply and fill the new environment. This is evident in bacteria, which are modern prokaryotes that can divide as rapidly as every 20 minutes. This allows a single individual to generate more than 4 billion descendants—approximately the human population of this planet—within 10 hours.
Cell cycling and DNA replication in eukaryotes (eukaryote = discrete nucleus), unlike that in prokaryotes, occur in spurts. The pauses between mitotic events allow the newly formed daughter cells to carry out their biological functions, replicate their DNA, and rebuild and reorganize their internal structure. The rate of cell cycling in eukaryotic cells varies greatly, pauses average 10 to 20 hours, but can be much longer. In adult human cardiac myocytes, most of which have lost their ability to divide, the pause can last until the host dies.
Nuclear division and cell division are usually tightly coordinated in cells that undergo mitosis, so that the cell cycle generates daughter cells that contain a single nucleus. In some cells, karyokinesis can occur without cytokinesis, so that the cell cycle generates a multinucleate cell. In other cells, DNA replication without either cytokinesis or karyokinesis generates polyploid cells whose nuclei contain more than two sets of chromosomes. In the normal adult human heart, ∼25% of cardiac myocytes are diploid, and more than half are tetraploid; the degree of ploidy increases with aging and when the heart hypertrophies (Rumyantsev, 1977).
Phases of the Cell Cycle
The cell cycle can be divided into four phases; in two, the cycle is active, in the other two it is relatively quiescent. The two active phases are the S phase, characterized by DNA synthesis and chromosome duplication, and the M phase, in which the duplicated chromosomes become separated and the cell divides (Fig. 9-2). These active phases are separated by two phases in which most processes related to cell division are temporarily suspended to allow cells to enlarge and carry out their physiological functions; these are the G1 phase, between M and S, and the G2 phase, between S and M. The G1 restriction point, which occurs late in G1, represents a major pause in cell cycling (Pardee, 1989). Once this restriction point is passed the cell is committed to dividing, which means that the cell cycle, while still governed by internal regulatory mechanisms, has become independent of external stimuli.
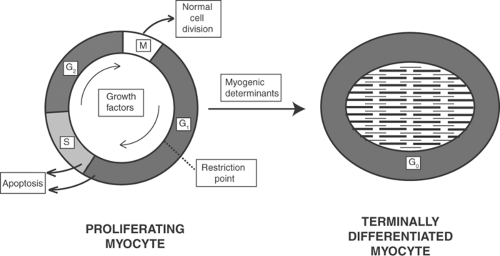 Fig. 9-2: Schematic depiction of the cell cycle and the transition to a terminally differentiated cell. Left: The cell cycle, such as occurs in proliferating fetal cardiac myocytes, proceeds in a clockwise direction. Mitosis (M) is followed by a “gap” phase (G1) during which cells enlarge and carry out their physiological functions; from the standpoint of the cell cycle this phase is quiescent. Cells become “committed” to the cell cycle when they pass a restriction point in G1, after which the cell cycle proceeds though a phase of DNA synthesis (S), a second gap phase (G2), and cell division (M). Cells are most susceptible to programmed cell death (apoptosis) late in the G1 phase and during the S phase. Right: Myogenic determinants cause cardiac myocytes to withdraw from the cell cycle and enter a prolonged quiescent phase of terminal differentiation, called G0. |
In terminally differentiated cardiac myocytes, the cells are in a G1 phase that resists stimuli which would otherwise activate cell cycling. This quiescent state, called G0 in this text, can be viewed as a “detour” out of the cell cycle. The term G0 is not strictly correct in describing this state in cardiac myocytes because G0 was initially coined to describe cell cycle arrest associated with low metabolic activity in serum-depleted cultured cells. A more accurate characterization of adult cardiac myocytes might be “permanent arrest in the G1 phase,” but with apologies to experts, this text uses the simpler term G0.
Adult cardiac myocytes have little or no ability to reenter the cell cycle, even when exposed to stimuli that would activate mitosis in less differentiated cells. As noted above, a few severely stressed cardiac myocytes can reverse the transition from G1 to G0, and so reenter the cell cycle, but these proliferative responses have virtually no ability to regenerate functioning cardiac myocytes.
Although proliferative signaling in mammalian hearts cannot increase cell number (hyperplasia), it does cause the myocytes to enlarge (hypertrophy). However, these efforts turn out badly because, in spite of increasing sarcomere number, hypertrophy of diseased or overloaded hearts generally has deleterious consequences that hasten progression of hemodynamic abnormalities, accelerate myocardial cell death, and shorten life expectancy (see Chapter 18).
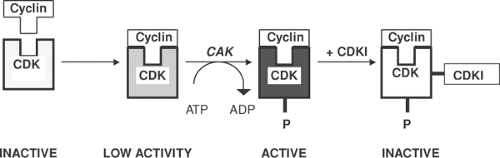 Fig. 9-3: Cyclin-dependent protein kinases (CDKs) regulate the cell cycle when they interact specifically with regulatory proteins called cyclins to form cyclin/CDK pairs. Most cyclins activate CDK activity only partially; full activation occurs when the cyclin-bound CDKs are phosphorylated by other protein kinases called CDK-activating kinases (CAK). Phosphorylated cyclin/CDK pairs are inactivated by cyclin-dependent kinase inhibitors (CDKIs). Additional regulators (not shown) include phosphatases that dephosphorylate the CDKs. |
Cyclins, CDK, and Related Proteins
The cell cycle is controlled by heterodimeric serine/threonine kinases that include catalytic subunits called cyclin-dependent kinases (CDKs) and regulatory subunits called cyclins that determine the substrate specificity of the CDK (Fig. 9-3). Cyclin/CDK pairs regulate transitions within and between the phases of the cell cycle shown in Figure 9-2; some stimulate cell growth in G1, others promote DNA replication in S, induce cell division in M, and stimulate transitions from G2 to M and from G1 to S. The cyclin D/CDK4 pair allows the cell cycle to pass the G1 restriction point by releasing the transcription factor E2F from an inhibitory complex with a hypophosphorylated retinoblastoma protein (see below). Cyclin/CDK pairs also regulate the activity of a variety of transcription factors.
CDKs are enzymatically inactive unless bound to an appropriate cyclin, but cyclins generally cause only partial activation of their corresponding CDKs. Complete activation often requires the participation of additional protein kinases, called CDK-activating kinases (CAK), that phosphorylate the cyclin/CDK pairs, while cyclin-dependent kinase inhibitors (CDKIs) slow the cell cycle by inhibiting active cyclin/CDK pairs (Fig. 9-3). Additional regulatory proteins, not shown in Figure 9-3, include phosphatases that inhibit cell cycling by dephosphorylating CDKs, and protein kinases that inactivate cyclin/CDK pairs by catalyzing inhibitory phosphorylations.
Retinoblastoma Proteins and E2F
Cell cycle progression is regulated by proteins whose prototype is the retinoblastoma protein (pRB); these proteins are sometimes referred to as tumor suppressors because they are substrates for phosphorylations that regulate cell differentiation and inhibit tumor formation, or pocket proteins because their structure includes a pocket that can bind transcription factors called E2F. Retinal cells that lack both copies of the pRB gene undergo malignant transformation and can form metastasizing tumors. Some retinoblastoma proteins are substrates for cyclin/CDK-mediated phosphorylations that regulate the cell cycle. In the heart, where the most important pRB is p107, the pRB and E2F regulate DNA repair and apoptosis, and can activate or inhibit gene expression.
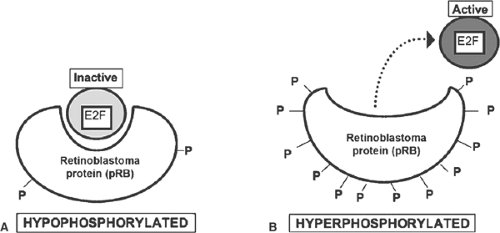 Fig. 9-4: Function of retinoblastoma proteins (pRBs). A: In the hypophosphorylated state, pRBs bind and inactivate transcription factors such as E2F. B: Phosphorylation of serine and threonine residues reduces the affinity of pRBs for these transcription factors, which are released in an active form that regulates proliferative signaling. |
In hypophosphorylated pRB, the pocket binds to and inactivates E2F, which arrests the cell cycle in the G1 phase. Hyperphosphorylation of serine and threonine residues causes pRB to activate E2F by “kicking” this transcription factor out of the pocket (Fig. 9-4). E2Fs can also be inactivated by phosphorylation or direct binding to the cyclin A/CDK2 pair (Fig. 9-5). Together, pRB and E2F provide a “switch” that controls passage of the cell cycle through the G1 restriction point.
A variety of signaling systems in addition to CDK/cyclin pairs regulate proliferative signaling by activating E2F. These include receptor tyrosine kinases, steroid receptors, G protein-coupled receptors, and cytoskeletal deformation. E2F activity can also be regulated by changes in the rate at which this transcription factor is synthesized, which allows increased levels of the messenger RNA (mRNA) that encodes E2F to stimulate proliferative signaling.
Genomic Regulation of Phenotype
Genomic mechanisms, which are among the most highly regulated of all biological functions, depend on the base pair sequence in chromosomal DNA to control phenotype by directing gene expression (Table 9-1). In normal hearts, most genes are down-regulated or dormant unless their expression is turned on by transcription factors. Once activated, genomic DNA serves as the template for the formation of primary RNA transcripts whose sequences are transcribed in the nucleus to form mRNA; the latter is then transported to the cytosol where it serves as a template for the amino acid sequences of proteins.
Rna Polymerases and Transcription Factors
Activation of gene expression begins when specific DNA sequences called promoters bind to multiprotein enzyme aggregates called RNA polymerases (Fig. 9-6). The latter operate in concert with general transcription factors that, along with the RNA polymerase, bind to the promoter.
DNA helicases that are included in the RNA polymerase then unwind a short segment of the two strands of base pairs, which allows the downstream DNA to serve as a template for the primary RNA transcript. The latter, which is synthesized by the RNA polymerase as it moves downstream along the gene, contains the information encoded in the DNA. Processing of the transcript then forms mRNA, which is transported to the cytosol where it determines the amino acid sequence in newly synthesized proteins.
DNA helicases that are included in the RNA polymerase then unwind a short segment of the two strands of base pairs, which allows the downstream DNA to serve as a template for the primary RNA transcript. The latter, which is synthesized by the RNA polymerase as it moves downstream along the gene, contains the information encoded in the DNA. Processing of the transcript then forms mRNA, which is transported to the cytosol where it determines the amino acid sequence in newly synthesized proteins.
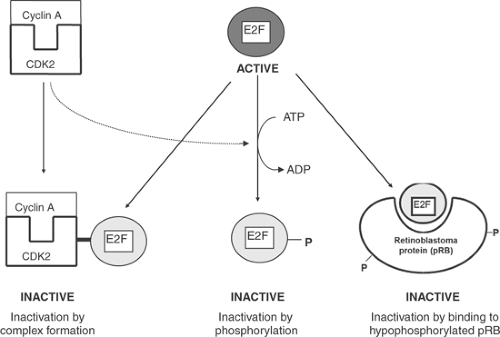 Fig. 9-5: E2F can be inactivated when it forms a complex with the cyclin/CDK pair cyclin A/CDK2 (left), by CDK-catalyzed phosphorylation (center), and when it binds to a hypophosphorylated retinoblastoma protein (right). CDK, cyclin-dependent kinase. |
In addition to the nucleotide sequences that direct protein synthesis, genomic DNA contains sequences that control the rate at which genes are transcribed, and turn genes on and off. These regulatory sequences include promoter regions, which are located at the 5′-end of the gene, “upstream” from the start site where DNA transcription begins, and enhancer and repressor regions that increase and decrease the rate of gene expression, respectively (Fig. 9-6). One of the most important promoter regions is the TATA box, so named because its DNA sequence includes thymidine (T) and adenine (A) in the sequence TATA; this sequence is a weak point in the double-stranded DNA where the helix is most easily unwound into the separate single strands used for copying. A remarkable discovery that was made possible by the human genome project is that a large majority of the information encoded in genomic DNA provides templates for small RNA segments, called miRNA, that control the epigenetic regulation of phenotype (see below). Basic proteins, called histones, which bind to and stabilize genomic DNA in chromatin, also participate in epigenetic regulation.
Transcription factors, most of which are proteins and phosphoproteins, turn genes on by binding to a promoter region, accelerate gene expression by binding to an enhancer sequence, or slow gene transcription by binding to a repressor sequence (see Fig. 9-6). The ability of transcription factors to select a specific gene and then regulate its expression is made possible by structural motifs that provide the tight “fit” needed to identify a specific target DNA sequence. Most operate
as homo- or heterodimers in helix-turn-helix, helix-loop-helix, leucine zipper, and zinc finger structures (Fig. 9-7), where each subunit binds to one of the two strands of genomic DNA. The subunits in these dimers are held together by non-covalent linkages that include hydrophobic interactions between leucine residues and divalent zinc atoms. Gene expression can also be regulated by hormones, such as thyroxin and aldosterone. These hydrophobic molecules cross the
plasma membrane and bind to intracellular nuclear receptors (also called hormone receptors); the resulting hormone-receptor complexes then serve as transcription factors that regulate gene expression. Cooperative interactions between genes and transcription factors can allow a single transcription factor to generate a coordinated growth response by interacting sequentially with several functionally related genes, and regulatory regions of single genes to modify the actions of several transcription factors.
as homo- or heterodimers in helix-turn-helix, helix-loop-helix, leucine zipper, and zinc finger structures (Fig. 9-7), where each subunit binds to one of the two strands of genomic DNA. The subunits in these dimers are held together by non-covalent linkages that include hydrophobic interactions between leucine residues and divalent zinc atoms. Gene expression can also be regulated by hormones, such as thyroxin and aldosterone. These hydrophobic molecules cross the
plasma membrane and bind to intracellular nuclear receptors (also called hormone receptors); the resulting hormone-receptor complexes then serve as transcription factors that regulate gene expression. Cooperative interactions between genes and transcription factors can allow a single transcription factor to generate a coordinated growth response by interacting sequentially with several functionally related genes, and regulatory regions of single genes to modify the actions of several transcription factors.
Table 9-1 Regulation of Gene Expression and Protein Composition in the Heart | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
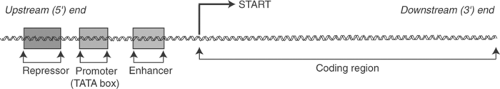 Fig. 9-6: Gene transcription. Coding regions of double-stranded genomic DNA that lie downstream (toward the 3′ end of the DNA sequence) from a “START” sequence provide templates for the primary RNA transcript that encodes messenger RNA. Gene transcription begins when RNA polymerases actively bind to a regulatory sequence located upstream from the 5′ end of the gene, called a promoter; the TATA box, which is made up of thymidine (T) and adenine (A) residues, is among the most important of these promoters. Transcription can be activated by enhancers and inhibited by repressors; the latter are found not only upstream from the start site, as shown in the figure, but also downstream and within regulatory sequences in the coding region. |
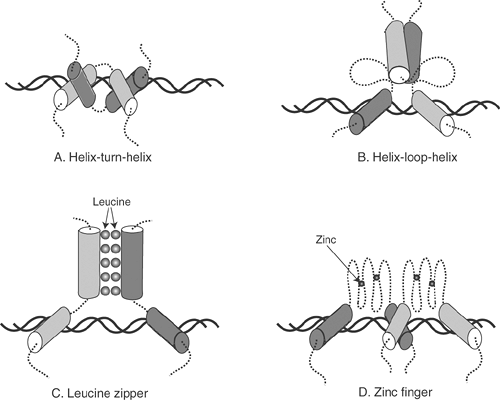 Fig. 9-7: Four types of transcription factor. These regulators, which function as dimers, include helix-turn-helix (A) and helix-loop-helix structures (B), leucine zipper structures (C) in which the two subunits of the dimer are linked by hydrophobic forces between leucine residues, and zinc finger structures (D) where zinc atoms stabilize the structure of the transcription factor. Cylinders represent α-helices. |
Transcription factors can be activated in many ways. Most common is posttranslational modification of the inactive protein, such as by phosphorylation or dephosphorylation. Transcription factor activity can also be regulated when another proliferative signaling cascade increases or decreases the rate of its synthesis, releases the active protein from an inactive complex, or incorporates the transcription factor into aggregates that are often called scaffolds.
The DNA strands at the ends of each chromosome are capped by telomeres. These structures, which in humans contain hundreds of copies of the tandem repeat TTAGGG along with members of a family of protective proteins called shelterins, protect the chromosomes from enzymes that normally degrade broken DNA strands. The number of tandem repeats in the telomere
decreases with normal aging, and DNA damage associated with telomere shortening has been suggested to contribute to the pathogenesis of heart failure.
decreases with normal aging, and DNA damage associated with telomere shortening has been suggested to contribute to the pathogenesis of heart failure.
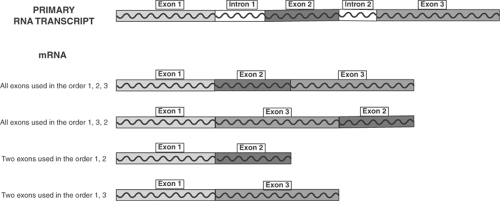 Fig. 9-8: Alternative splicing. During processing of the primary RNA transcript, the sequences in exons are encoded in proteins, while those in introns are removed prior to synthesis of messenger RNA (mRNA). Selection and rearrangement of exons allows a single gene to encode several different mRNAs. In this example, three exons in the primary transcript form four mRNAs by alternative splicing. These are only a few of the possible mRNAs in which one, two or all three exons can be combined in various sequences. |
Rna Processing and Alternative Splicing
Some segments of the primary transcript, called introns, are eliminated before the remaining nucleotide sequences, called exons, are assembled into the mRNA that directs protein synthesis. Elimination of introns and rearrangement of selected exons, called alternative splicing, is an important mechanism that provides diversity in proliferative signaling. Figure 9-8 shows how a gene containing three exons can encode four different mRNAs; this illustration is by no means complete as the three exons can be used in additional combinations. Some genes contain dozens of exons, so that alternative splicing can generate dozens of protein isoforms from a single DNA sequence.
Epigenetic Regulation
In addition to the genomic mechanisms described above, which select specific genes for expression or suppression and allow alternative splicing to modify gene transcripts, a recently discovered group of regulatory mechanisms regulates proliferative signaling. The growing impact of these mechanisms, called epigenetics, is reminiscent of the late 1950s, when as a research fellow in C. B. Anfinsen’s laboratory at the National Institutes of Health in Bethesda Maryland, I heard Francis Crick describe the beginning of the DNA revolution (the double helix, genetic code, etc.). Today’s emergence of epigenetics promises another biological revolution that is likely to be even more complex than genetics.
Defined most broadly, epigenetics refers to non-Mendelian mechanisms of inheritance that influence phenotype without depending directly on the sequence of the base pairs in DNA. Genomics and epigenetics can be contrasted by viewing the former as using information encoded in genomic
DNA to determine how cells, organs, and overall body plan can appear, whereas epigenetic mechanisms determine how this genetic information is used to determine what does appear.
DNA to determine how cells, organs, and overall body plan can appear, whereas epigenetic mechanisms determine how this genetic information is used to determine what does appear.
Among the more remarkable features of epigenetics is that regulation by RNA can explain why, although more than 93% of the DNA in the human genome is transcribed, less than 2% of the RNA transcript encodes proteins. This, of course, raises the question: “What happens to the non-protein-coding RNA transcript?” The answer is that many of these RNA transcripts regulate gene expression. It is amazing, although there is ample precedent in many other fields of science, that for a half century the importance of RNA in regulating gene expression was overlooked almost completely.
An unexpected feature of epigenetic regulation is that it can allow environmental factors, such as nutritional state, temperature, toxins, and even maternal nurturing, to induce changes in phenotype that can be passed down to successive generations (Jirtle and Skinner, 2007). The ability of epigenetic changes to modify the germ line marks a return toward the 19th century Lamarckian view that environment can modify inherited traits.
In cardiology, epigenetic mechanisms help regulate impulse conduction, contribute to molecular changes in failing hearts, cause several inherited cardiomyopathies, and influence the clinical manifestations of specific gene mutations. The influence of epigenetic mechanisms on phenotypic expression also explains variations in the severity of the clinical syndromes associated with specific genomic mutations, often called penetrance (see, for example, Zhou et al., 2006), and how a single genotype can give rise to different phenotypes (Fraga et al., 2005).
Three types of mechanisms participate in epigenetic control (Table 9-2). The first includes changes in DNA brought about by mobile genetic elements, covalent modification of genomic
DNA by cytosine methylation, and interactions between trans-gene alleles. In the second, small sequences of RNA, called RNAi, inhibit (“silence”) RNA translation; the third, which modifies chromosome and chromatin structure, includes posttranslational covalent modifications of histone that alter the ability of genomic DNA to interact with transcriptional regulators.
DNA by cytosine methylation, and interactions between trans-gene alleles. In the second, small sequences of RNA, called RNAi, inhibit (“silence”) RNA translation; the third, which modifies chromosome and chromatin structure, includes posttranslational covalent modifications of histone that alter the ability of genomic DNA to interact with transcriptional regulators.
Table 9-2 Some Epigenetic Mechanisms | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Dna Modification
More than 50 years ago, McClintock (1950) found that a given gene could appear at various loci in the maize genome. This genetic instability, which was initially referred to as “jumping genes,” reflects the movement of DNA sequences between non-homologous regions of the genomic DNA. The mobile DNA segments, called tranposons, range in size from hundreds to tens of thousands of base pairs. A related epigenetic mechanism, called parental or genomic imprinting or parent-of-origin regulation, can inactivate large segments of either X or Y chromosomes.
DNA methylation, which generally “locks” genes in a silent state, is among the most ancient of the epigenetic mechanisms. It probably appeared as a defense mechanism that allowed unwilling hosts to eliminate invading parasites by methylating, and so inactivating, parasite genomes. In eukaryotes, DNA methyl transferases, which catalyze the addition of methyl groups to cytosine, inhibit gene transcription and apoptosis. Demethylation, which is catalyzed by DNA demethylases, activates proliferative signaling, while methyl-CpG binding proteins identify and initiate the methylation of specific DNA sequences. Cytosine methylation has been linked to destabilization of dinucleotide 5′-CpG-3′ (CpG) tandem repeats and contributes to the pathogenesis of several human diseases (Robertson, 2005; Rodenhiser and Mann, 2007).
Interactions between trans-regions of paired or homologous DNA sequences can silence gene expression by inhibiting the transcription and/or translation of specific genes. These responses, called paramutation, are regulated by such epigenetic mechanisms as DNA methylation and RNA inhibition.
Rna Modifications
RNA interference (RNAi), which is emerging as one of the most important epigenetic mechanisms that determine phenotype, occurs when a short (19 to 25 nucleotide), single-stranded antisense RNA sequence, called a miRNA, modifies gene expression. At the end of 2009, more than 700 different human miRNAs were mentioned in the Sanger miRBase (http://www. mirbase.org/). miRNAs regulate many functions in the heart such as myogenesis, cardiac development, and proliferative responses that include maladaptive features of the heart’s response to hemodynamic overload. miRNAs have been found to be upregulated and downregulated in failing hearts, and a distinct abnormal miRNA expression pattern has been reported in human-dilated cardiomyopathies (Naga Prasad et al., 2009).
Synthesis of miRNAs (Fig. 9-9) begins when pri-RNA, one of the primary transcripts that do not encode proteins (see above), is cropped in the nucleus by an endonuclease called drosha. This forms ∼70 nucleotide pre-miRNAs that are transported to the cytoplasm where they are cleaved by another endonuclease called dicer. This releases a shortened double-stranded RNA (RNA duplex) that aggregates with several proteins to form an RNA-induced silencing complex (RISC). One of the proteins that make up the RISC, called argonaute, hydrolyzes the double-stranded RNA duplex to liberate the single-stranded miRNAs that bind with high specificity to mRNA sequences. miRNAs can inhibit the synthesis of specific proteins, catalyze the degradation of other mRNA sequences, and even reenter the nucleus where they can serve as transcription factors. Artificial miRNAs called small interfering (si)RNAs, which can be synthesized in vitro, can also silence specific genes.
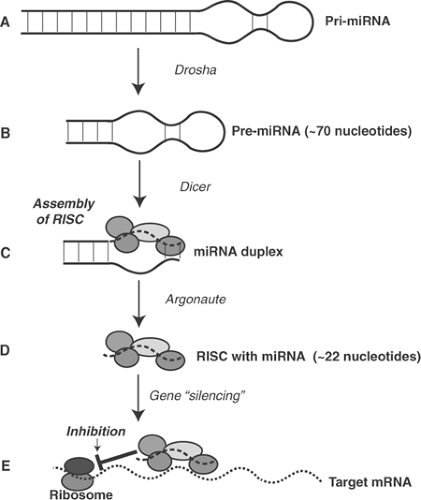 Fig. 9-9: microRNA synthesis. A: MicroRNA precursors, called pri-miRNA, are transcribed from genomic DNA in the nucleus. B: Drosha, an endonuclease, cleaves ∼70 nucleotide double-stranded pre-MiRNA from the pri-miRNA. C: The pre-miRNA is processed by dicer to form a duplex that contains a shortened RNA, one strand of which will become the miRNA (dashed line). D: The double-stranded pre-miRNA forms RNA-induced silencing complexes (RISC) with several regulatory proteins that include argonaute, which releases the short, ∼22 nucleotide, singe-stranded miRNA. E: Binding of RISCs to ribosomal RNA sequences (target mRNA) allows the miRNA to silence (inhibit) translation of the latter. mRNA, messenger RNA. |
Chromosomal Changes
Chromosomes are stabilized in structures called nucleosomes in which the two strands of DNA are wound tightly around histone cores, which causes the genomic DNA to become inaccessible to transcription factors. Acetylation, methylation, phosphorylation, ubiquitination, and ribosylation of the á-amino lysyl groups in histone can activate gene transcription by unwinding the nucleosomes. Histone acetylation by histone acetyltransferases (HATs) stimulates proliferative signaling by exposing active sites on the DNA (Fig. 9-10). Conversely, removal of the acetyl groups by histone deacetylases (HDACs) reverses this stimulatory effect by causing DNA to condense in chromatin where it becomes inaccessible to transcriptional regulation. The activity of HATs and HDACs is controlled by a number of signaling cascades. For example, phosphorylation of some HDACs by calcium-calmodulin (CAM) kinases and protein kinase C (PKC) increases their ability to acetylate histones, which inhibits responses to transcription factors that lead to pathological hypertrophy.
Gene expression is also regulated by epigenetic mechanisms that alter the spatial organization of genes and transcriptional regulators within the nucleus. This can occur when specific regions of the genomic DNA are compartmentalized in specialized chromatin loops when chromosomes are arranged in various degrees of proximity to one another at the periphery of the nucleus, and when chromosome positions change during anaphase.
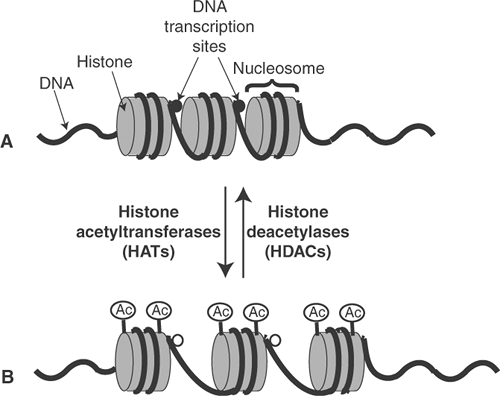 Fig. 9-10: Epigenetic regulation of DNA transcription by histone acetylation. A: Deacetylated histone forms tightly packed nucleosomes in which DNA transcription sites (closed circles) cannot interact with transcription factors. B: Acetylation of histone by histone acetyltransferases (HATs) inhibits chromatin condensation by unwinding histone-bound DNA, which allows the DNA transcription sites (open circles) to interact with transcription factors. The activated DNA transcription sites are inactivated when histone is deacetylated by histone deacetylases (HDACs). |
Extracellular Growth Factors
Many proliferative signaling mechanisms that operate within cells are controlled by extracellular messengers. The importance of the latter became apparent in the middle of the 20th century, when adult cells cultured in artificial media were found to lose their ability to grow and divide even after addition of all known nutrients, vitamins, and trace elements. The key to these puzzling observations was provided by the observation that inclusion of fetal calf serum in the artificial media allowed the cultured cells to proliferate. Efforts to characterize the unknown factors needed to maintain cell cycling identified the missing ingredients as peptides that came to be called growth factors. Several of these peptides, along with cytokines and most of mediators of the functional responses described in Chapter 8, are now known to modify proliferative signaling (Table 9-3).
Peptide Growth Factors
A large number of peptides stimulate proliferative responses. Although initially named for the tissues from which they were first isolated, or whose growth they were initially found to stimulate, most peptide growth factors turned out not to be tissue-specific, but instead are able to mediate proliferative signaling in many cell types throughout the body. These include platelet-derived growth factor (PDGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF), and vascular endothelial growth factor (VEGF), along with transforming growth factors (TGF) and other cytokines (Table 9-4). All use paracrine, autocrine, and endocrine pathways to reach receptors on their target cells, most of which contain a latent tyrosine kinase that is activated when the receptors form aggregates with their ligands. A few, like TGF-β receptors, have latent serine/threonine kinase activity. Cytokine receptors, which lack an intrinsic protein kinase, activate latent tyrosine kinase activity in other membrane-associated proteins with which they form aggregates.
Two fundamentally different mechanisms allow binding of a growth factor to its receptor to activate intracellular signal transduction cascades (Fig. 9-11). In the first, the ligand-receptor
complex activates a protein kinase or other enzyme that then modifies an intracellular signaling molecule (Fig. 9-11A). In the second, binding of the growth factor to its receptor initiates the formation of a multiprotein aggregate containing a docking site (scaffold) that binds to additional intracellular signaling molecules (Fig. 9-11B). Activation of proliferative signaling by internalized β2 receptors (see Chapter 8) is one example of the latter mechanism; other examples are described below.
complex activates a protein kinase or other enzyme that then modifies an intracellular signaling molecule (Fig. 9-11A). In the second, binding of the growth factor to its receptor initiates the formation of a multiprotein aggregate containing a docking site (scaffold) that binds to additional intracellular signaling molecules (Fig. 9-11B). Activation of proliferative signaling by internalized β2 receptors (see Chapter 8) is one example of the latter mechanism; other examples are described below.
Table 9-3 Some Proliferative Signaling Systems That Operate in the Mammalian Cardiovascular System | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


