Signal Transduction Pathways of the Heart
Johannes Backs
Eric N. Olson
Rhonda Bassel-Duby
Overview
Signal transduction is a cascade of processes by which an extracellular signal interacts with a receptor at the cell surface, causes a change in the level of a second messenger, and ultimately alters gene expression and the phenotype of the cell. After birth, terminally differentiated cardiac myocytes respond to neurohormonal stimulation primarily by activating membrane-bound receptors, thus causing intracellular signaling through kinases, phosphatases, and guanosine triphosphatase (GTPase) proteins and promoting changes in cardiac gene expression and cellular phenotypes. Many of these cellular responses increase protein synthesis, enlarge cell size, and augment the organizational structure of the sarcomere, with resulting cardiac hypertrophy. Physiologic cardiac hypertrophy, considered normal growth of the heart postnatally, occurs in response to conditioning exercise and enhances cardiac output to meet the increased metabolic demand. In contrast, pathologic hypertrophy occurs in response to biomechanical and hemodynamic stresses and triggers a metabolic transition in the heart from an oxidative toward a more glycolytic metabolism characteristic of the fetal stage. In addition, pathologic hypertrophy is accompanied by the activation of a fetal cardiac gene program that includes genes whose products regulate cardiac contractility and calcium handling (1). Although cardiac hypertrophy initially is considered an adaptive response to injury by normalizing wall stress and maintaining normal cardiac function, prolonged hypertrophy can lead to diastolic and, later, systolic heart failure and cardiac sudden death from arrhythmias. Whether physiologic and pathologic forms of hypertrophy are controlled by the same or different pathways is a central issue in the field with significant clinical implications because enhancing the former and suppressing the latter could have profound therapeutic consequences. This chapter highlights various signaling pathways shown to be involved in cardiac growth, with the objective of identifying molecular factors that could be targeted for drug development to ameliorate heart disease.
Much of what we know about the signaling pathways in the heart has been discovered by gain-of-function or loss-of-function approaches using genetic engineering to generate transgenic mouse models of heart disease. Using insertional genetic manipulations, signal transduction cascades are activated in the heart by the introduction of an active form of a signaling protein using a cardiac-specific promoter. These transgenic mice models serve as indicators of a cardiac phenotype elicited by activation of a particular signaling pathway. Conversely, using transgenic mouse models to express specific inhibitors in the heart provides an approach to observe the effects when signal transduction pathways are blocked in vivo. Targeted gene deletion by homologous recombination evaluates the cardiac phenotype resulting from the loss of a specific factor in the signaling pathway. These genetically modified mouse models provide the preliminary observations implicating the involvement of a molecular determinant in a signal transduction pathway effecting cardiac remodeling. It still remains to be determined whether these factors and signaling pathways are pertinent to human heart disease.
G-Protein–Coupled Receptor Signaling Pathway
Activation of neurohormonal systems plays a major role in the progression of heart failure. β-Blockers and angiotensin-converting enzyme inhibitors have been shown to improve parameters such as ventricular remodeling, ejection fraction, and renal function and to reduce rates of morbidity and mortality. Likewise, G-protein–coupled receptors (GPCRs), which are the receptors for many important neurohormones, play important roles in the regulation of cardiac function and adaptation to changes in hemodynamic burden (2). GPCRs are coupled to three principal classes of heterotrimeric GTP-binding proteins, Gq/G11, Gs, and Gi, which transduce agonist-induced signals towards intracellular effectors such as enzymes and ion
channels. All heterotrimeric G proteins consist of the subunits Gα and Gβγ, which on receptor activation dissociate and independently activate intracellular signaling pathways.
channels. All heterotrimeric G proteins consist of the subunits Gα and Gβγ, which on receptor activation dissociate and independently activate intracellular signaling pathways.
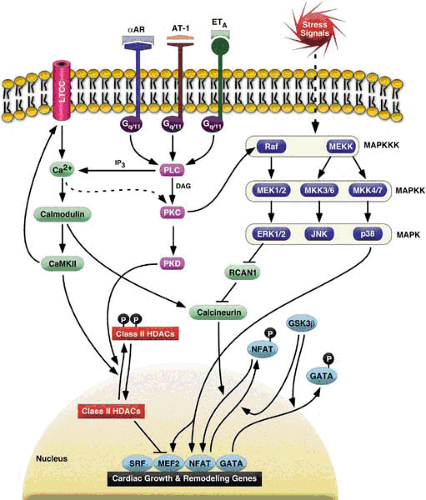 FIGURE 99.1. G-protein–coupled receptor signaling pathway. Neurohormones and stress signals activate the G-protein–coupled receptor pathway by transducing intracellular factors and activating cardiac remodeling genes. In the mitogen-activated protein kinase (MAPK) signaling pathway, activation of MAPK kinase kinases (MEKKs and Raf) activates MAPK kinases (MEK1/2, MKK3/6, and MKK4/7) and facilitates binding and direct phosphorylation of extracellularly responsive kinase 1/2 (ERK1/2), c-Jun N-terminal kinase 1/2/3 (JNK1/2/3), and p38. αAR, α-adrenergic; AT-1, angiotensin-1 receptor; CaMKII, calcium/calmodulin–dependent protein kinase; DAG, diacylglycerols; ETA, endothelin A receptor; GATA, GATA binding protein; GSK3β, glycogen synthase kinase 3β; HDAC, histone deacetylase; IP3, inositol 1,4,5-triphosphate; LTCC, L-type calcium channel; MEF2, myocyte enhancer factor-2; NFAT, nuclear factor of activated T cells; PLC, phospholipase C; PKC, protein kinase C; PKD, PKCμ/protein kinase D; RCAN1, regulator of calcineurin-1; SR, sarcoplasmic reticulum; SRF, serum response factor. |
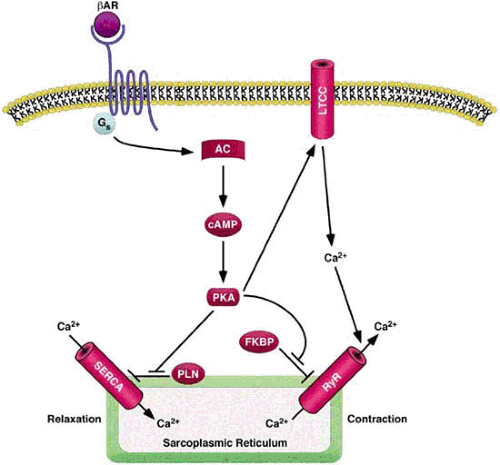 FIGURE 99.2. Signaling pathway calcium handling. Calcium (Ca2+) enters the cardiac muscle cell through L-type calcium channels (LTCC) and triggers further calcium release from the sarcoplasmic reticulum through the ryanodine receptor (RyR), thus raising the free intracellular calcium concentration. Activation of the β1-adrenergic receptor (βAR) signals protein kinase A (PKA) to phosphorylate phospholamban (PLN), there by enhancing sarcoplasmic reticulum calcium adenosine triphosphatase (SERCA) activity and promoting cardiac muscle relaxation. AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; FKBP, FK-506 binding protein. |
Endothelin-1 (ET-1), angiotensin II (Ang II), and norepinephrine bind their receptors (ET-A, AT-1, and α-adrenergic), which are coupled to Gq/11 and which, in turn, activates phospholipase C (PLC) and results in sustained elevated cytoplasmic calcium levels and activation of protein kinase C (PKC) (Fig. 99.1). These agonists have all been shown to induce cardiomyocyte hypertrophy (3). Mice lacking dopamine ß-hydroxylase, an essential enzyme for the synthesis of norepinephrine, exhibit a blunted hypertrophic response with preserved contractility after aortic banding (4). Mice overexpressing GPCRs (5,6) as well as their downstream mediator Gq (7,8) develop cardiac hypertrophy and subsequently depressed contractile function. Conversely, combined ablation of the genes encoding for Gq and G11 results in embryonic lethality caused by myocardial hypoplasia, a finding again suggesting an important role of these mediators in the control of cardiac growth (9). G11-null mice, which also lack Gq specifically in the heart, do not develop cardiac hypertrophy or activation of the fetal gene program in response to aortic banding (10), a finding demonstrating a requirement for Gq/G11 for most, if not all features, of pressure overload–induced cardiac hypertrophy. The clinical observation that treatment of patients with heart failure with AT-1 receptor blockers and angiotensin-converting enzyme inhibitors inhibits cardiac remodeling supports these findings (11).
The most abundant adrenergic receptor in cardiac tissue is the β1-adrenergic receptor, which is coupled to Gs and, in turn, activates adenylate cyclase (AC) (Fig. 99.2). The less abundant β2-adrenergic receptor can couple to both Gs and Gi. Mice overexpressing the β1-adrenergic receptor in the heart initially show increased contractile function and responsiveness to isoproterenol, but eventually they demonstrate deterioration of cardiac performance, cardiomyocyte hypertrophy, and fibrosis (12,13). Transgenic cardiac overexpression of Gs resulted in a similar phenotype, but surprisingly it did not depend on activation of AC (14). In contrast, overexpression of AC type VI does not appear to exert adverse effects on cardiac function and has been reported to attenuate cardiomyopathic changes, including cardiac hypertrophy in Gq transgenic mice (15,16). However, transgenic overexpression of protein kinase A (PKA), the principal target of AC, results in dilated cardiomyopathy associated with cardiomyocyte hypertrophy and fibrosis, a finding suggesting that PKA mediates the detrimental consequences of chronically elevated ß-adrenergic signaling, and AC may have other yet unknown targets that may confer cardioprotective effects (17). In contrast to β1-adrenergic signaling, cardiac overexpression of β2-adrenergic receptors is deleterious only at excessive levels, whereas moderate levels of β2-adrenergic receptor expression improve basal contractile function and rescue the cardiomyopathic phenotype of Gq-transgenic mice (18). Heart failure is accompanied by impaired ß-receptor function resulting from both a decreased number of receptors and functional uncoupling (19). The latter is mediated by β-adrenergic receptor kinase (ßARK) 1, which phosphorylates the receptor and thereby rapidly decreases its sensitivity to further agonist stimulation (20).
Clinical observations provide indirect evidence that the activation of β1-receptors plays an important role in the pathogenesis of cardiac hypertrophy and heart failure. Treatment of patients with heart failure with β-blocker drugs results in a rescue of the cardiomyopathic phenotype and alterations of gene expression, including downregulation of hypertrophic genes and upregulation of sarcoplasmic reticulum calcium adenosine triphosphatase (ATPase) (SERCA) 2a and α-myosin heavy
chain (MHC). The genetic reprogramming accompanying administration of β-blocker drugs may contribute to the beneficial effects observed with treatment (21).
chain (MHC). The genetic reprogramming accompanying administration of β-blocker drugs may contribute to the beneficial effects observed with treatment (21).
Both cardiac muscarinic and ß2-adrenergic receptors couple through Gi, thus inhibiting AC and directly opposing Gs-dependent signaling. Gi is upregulated in human heart failure (22,23), and basal AC activity is impaired, findings suggesting that this mechanism may contribute to the cardiomyopathic phenotype. Moreover, cardiac overexpression of a Gi-coupled GPCR resulted in cardiomyopathy and lethal arrhythmias (24), a finding implying that Gi-dependent signaling is sufficient to cause heart failure.
Calcium Handling
Calcium plays a central role in the control of cardiac growth, contractility, and function (25). Thus, it is not surprising that abnormalities in calcium handling have been implicated in many forms of adult cardiac disease. During each heartbeat, calcium enters the cardiac muscle cell through L-type calcium channels (Fig. 99.2). The increase in intracellular calcium triggers further calcium release from the sarcoplasmic reticulum through the ryanodine receptor (RyR), thus raising the free intracellular calcium concentration approximately 10-fold (26). Binding of calcium to troponin C in the contractile apparatus initiates muscle contraction. Reuptake of calcium into the sarcoplasmic reticulum by SERCA allows for cardiac relaxation. The ability of SERCA to pump calcium back into the sarcoplasmic reticulum is governed by its interaction with phospholamban (PLN), a small modulatory protein within the membrane of the sarcoplasmic reticulum (27). In the unphosphorylated state, PLN inhibits calcium uptake by SERCA. Signaling by PKA leads to phosphorylation of PLN, which diminishes its inhibitory activity, thereby allowing for enhanced activity of SERCA and promoting cardiac relaxation.
The RyR is a tetrameric channel that associates with multiple signaling molecules, including the FK-506 binding protein FKBP12.6, PKA, and protein phosphatases 1 and 2A (28). Interaction with FKBP12.6 stabilizes the channel in a closed state and reduces its activity. Phosphorylation of the RyR by PKA results in dissociation of FKBP12.6 and increased sensitivity to calcium-induced activation. Mice lacking FKBP12.6 or humans with mutations in the gene encoding the RyR that reduce its affinity for FKBP12.6 have “leaky” calcium channels with an increased probability of being open as well as an elevated susceptibility to exercise-induced cardiac arrhythmias (29). The RyR has also been reported to be hyperphosphorylated by PKA in failing human hearts, a feature that could potentially contribute to associated abnormalities in calcium handling and contractility.
In the failing heart, sarcoplasmic reticulum calcium uptake and release by the sarcoplasmic reticulum are diminished by a decrease in SERCA activity, and contractile performance is lost. The notion that aberrant calcium handling contributes to pathogenesis of the failing heart was supported by the finding that the heart failure phenotype of mice lacking the cardiac LIM domain protein MLP was completely abrogated by homozygous deletion of the PLN gene, which apparently allowed for enhanced calcium reuptake by the sarcoplasmic reticulum (30). Similarly, overexpression of SERCA can preserve cardiac function in certain mouse models of heart failure (31). These findings catalyzed interest in the possibility of introducing dominant-negative PLN into the failing heart through gene therapy, as a means of restoring sarcoplasmic reticulum calcium uptake and contractility. However, subsequent studies reported that the apparent salutary effects of PLN ablation are observed only in a subset of mouse models of heart failure (32). In humans, the recent discovery of PLN loss-of-function mutations in families with hereditary heart failure raised significant concerns about such an approach and underscored the potential pitfalls in directly extrapolating findings from the mouse to the human heart (33,34). The mere fact that the heart rates in mice and humans differ by an order of magnitude points to fundamental differences in cardiac control mechanisms in the two species. Nevertheless, it is obvious that functional abnormalities of SERCA, PLN, or both are central to the pathogenesis of heart failure.
Protein Kinase C
When activated by biomechanical or neurohormonal stress, Gq and G11 activate PLC, which, in turn, leads to an inositol 1,4,5-triphosphate (IP3)–mediated increase in cytosolic calcium and production of diacylglycerols (Fig. 99.1). One important effector arm of this cascade is the PKC family. PKC is a ubiquitously expressed serine/threonine kinase. Multiple studies implicate a role for PKC in the pathogenesis of cardiac hypertrophy. In the heart, the most abundant PKC isoenzymes belong to the “conventional” group (PKCα and PKCβ; calcium and diacylglycerol activated) and the novel group (PKCδ and PKCε; diacylglycerol activated but calcium independent). Phorbol esters and diacylglycerol analogs mimic phenylephrine-mediated cardiomyocyte hypertrophy (35). Overexpression of PKCß in transgenic hearts is sufficient to elicit cardiac hypertrophy and sudden death (36). However, these findings could not be confirmed in other studies (37,38). Moreover, PKCß is not necessary for the hypertrophic response to pressure overload or phenylephrine infusion, respectively, because targeted ablation of the PKCß gene in mice leaves the animal still susceptible to these stimuli (39). However, in isoform-specific PKC genetic ablation studies, the presence of other PKC isoenzymes in the heart may serve to be functionally redundant. PKCε has also been shown to cause compensated cardiac hypertrophy in vivo (40), whereas a similar study reported hypertrophy with rapid progression to heart failure (41). Utilizing dominant-negative adenoviral expression constructs, it was shown that only PKCα is both required and sufficient for cardiomyocyte hypertrophy in vitro (42). In vivo, hearts of mice lacking PKCα are hypercontractile and are protected against heart failure induced by pressure overload (43). Conversely, hearts of transgenic mice overexpressing PKCα are hypocontractile (43), a finding indicating a central role of PKCα in the regulation of cardiac contractility.
Evidence indicates that PKC is involved in myocardial ischemia. Both novel isoenzymes PKCε and PKCδ were shown to be upregulated in prolonged myocardial ischemia (44). Targeted disruption of PKCε blocked cardioprotection caused by ischemic preconditioning and α1-adrenergic receptor stimulation, a finding implicating PKCε as an essential component of cardioprotective signaling (45). Mice lacking PKCδ fail to benefit from ischemic preconditioning and display enhanced injury following ischemia and reperfusion. Consistent with these findings, no increase in reactive oxygen species was observed in PKCδ-null hearts, whereas it is recognized that ischemic preconditioning increases superoxide anion production in hearts of wild-type littermates (46,47).
Mitogen-Activated Protein Kinase Signaling Pathway
Mitogen-activated protein kinase (MAPK) pathways provide an important link between external stimuli and the nucleus via phosphorylation and regulation of multiple transcription factors (Fig. 99.1). Based on sequence homology, MAPKs can be divided into three major subfamilies, the extracellularly
responsive kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and the p38 MAPKs. The last two groups are also categorized as “stress-responsive” MAPKs because they can be activated not only by anabolic stimuli and agonists of GPCRs, but also by pathologic stress, such as ischemia or cytotoxic agents (48). Overexpression of MAPK phosphatase 1 (MKP-1), which inhibits all three major branches of MAPK signaling, blocked both agonist-induced hypertrophy in vitro and pressure overload–associated hypertrophy in vivo (49), thus demonstrating a significant role for these pathways in hypertrophic signaling. Moreover, fibroblast growth factor- (FGF-2)–null mice lack a hypertrophic response to renal hypertension, which is associated with a general defect in MAPK signaling, as demonstrated by significantly blunted activation of ERKs, JNKs, and p38 MAPKs (50). These effects were demonstrated to depend on paracrine FGF-2 release from cardiac fibroblasts, a finding suggesting an important role of these cells in mediating cardiac hypertrophy in a MAPK-dependent fashion.
responsive kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and the p38 MAPKs. The last two groups are also categorized as “stress-responsive” MAPKs because they can be activated not only by anabolic stimuli and agonists of GPCRs, but also by pathologic stress, such as ischemia or cytotoxic agents (48). Overexpression of MAPK phosphatase 1 (MKP-1), which inhibits all three major branches of MAPK signaling, blocked both agonist-induced hypertrophy in vitro and pressure overload–associated hypertrophy in vivo (49), thus demonstrating a significant role for these pathways in hypertrophic signaling. Moreover, fibroblast growth factor- (FGF-2)–null mice lack a hypertrophic response to renal hypertension, which is associated with a general defect in MAPK signaling, as demonstrated by significantly blunted activation of ERKs, JNKs, and p38 MAPKs (50). These effects were demonstrated to depend on paracrine FGF-2 release from cardiac fibroblasts, a finding suggesting an important role of these cells in mediating cardiac hypertrophy in a MAPK-dependent fashion.
Significant controversy surrounds the potential role of ERK1/2 in hypertrophic signaling. Although it was reported that depletion of ERK1/2 with antisense oligonucleotides or pharmacologic inhibition of MEK1/2 attenuated the hypertrophic response to agonist stimulation in cultured cardiomyocytes (51,52), other investigators could not confirm an inhibition of atrial natriuretic factor (ANF) induction when ERK1/2 signaling was inhibited (53). Moreover, ERK1/2 activation is not observed in transgenic hearts expressing Gq (8). However, in vivo data strongly support the notion that ERK1/2 dependent signaling is sufficient to evoke a hypertrophic phenotype. Transgenic overexpression of MEK1, an MAPK kinase that activates ERK1/2 but not JNKs or p38 MAPKs, results in considerable cardiac hypertrophy (54). In contrast to most hypertrophy models, this phenotype was associated with supernormal systolic function, whereas the hypertrophic gene program was induced in fashion similar to that seen in other “pathologic” hypertrophy models, a finding suggesting that MEK1-dependent signaling may thus constitute a pathway for “physiologic” hypertrophy.
MAPKs of the JNK class are directly phosphorylated by either MKK4 or MKK7, which, in turn, is regulated by MEKK1 phosphorylation. In cardiomyocytes, mechanical stretching (55) or agonist stimulation by ET-1, phenylephrine, or Ang II results in rapid phosphorylation of JNK (56,57,58). Moreover, MKK7 has been shown to be sufficient to induce all features of cardiomyocyte hypertrophy when it is overexpressed in cultured cardiomyocytes (59). Conversely, adenovirus-mediated expression of a dominant-negative MKK4 mutant attenuates the hypertrophic response to ET-1 in vitro (55), as well as pressure overload–induced hypertrophy (60). Further evidence for an important role of JNK signaling in cardiac hypertrophy stems from studies in mice with a targeted disruption of the MEKK1 gene, which results in selective attenuation of JNK activity. MEKK1/JNK has been implicated in the hypertrophic response of cardiomyocytes secondary to Gq-coupled receptor stimulation (61). Accordingly, in mice both deficient for MEKK1 and transgenic for Gq, the hypertrophic phenotype, as well as JNK activation of the latter model, is entirely abrogated (62). Moreover, this effect was associated with improved systolic function, a finding suggesting that MEKK1 is necessary for most, if not all, adverse consequences of chronic Gq activation in the heart. In contrast, MEKK1 deficiency not only failed to prevent pressure overload hypertrophy related to aortic banding, but also even resulted in accelerated progression to dilated cardiomyopathy (63), a finding suggesting that MEKK1/JNK inhibition is not a generally applicable strategy to prevent the adverse consequences of cardiac hypertrophy.
The most important activators of p38 MAPKs are MKK3 and MKK6, both of which are sufficient to induce cardiomyocyte hypertrophy and ANF induction in vitro (64). Similar to the other branches of MAPK signaling, p38 activity is induced in response to pressure overload (65) and agonist stimulation (51,66). In addition, TAK1, which is upstream of MKK3/6, is upregulated and is activated in vivo after aortic banding (67). A cardiac-specific, constitutively active TAK1 mutant results in cardiac hypertrophy and subsequent heart failure in transgenic mice, a finding further implicating this branch of the MAPK-signaling pathway in pathologic growth of the myocardium (67). Interestingly, p38 phosphorylates several transcription factors in hypertrophic gene expression, including myocyte enhancer factor-2 (MEF2) and nuclear factor of activated T cells-3 (NFAT3) (see later) (68,69).
Calcineurin/Nuclear Factor of Activated T cells Signaling Pathway
The calcium-dependent, serine/threonine phosphatase calcineurin (also called protein phosphatase 2B) is a critical transducer of calcium signals that govern cardiac adaptation and disease (70,71,72




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
