Signal Transduction: Functional Signaling
The vie constante, where life manifests itself independently of the external environment [is] characterized by freedom and independence … Here life is never suspended, but flows steadily on apparently indifferent to alterations in its cosmic environment or changes in its material surroundings. Organs, structural mechanisms and tissues all function uniformly … [Because] the milieu intárieur surrounding the organs, the tissues, and their elements never varies; atmospheric changes cannot penetrate beyond … we have an organism which has enclosed itself in a kind of a hot-house. The perpetual changes of external conditions cannot reach it; it is not subject to them, but is free and independent.
—Claude Bernard, 1878
Bernard (1878) noted that constancy of the milieu intárieur is essential for independent life. Maintenance of this internal environment, which Cannon (1932) called homeostasis, is made possible by mechanisms that compensate for changes in the labile, often hazardous, external environment. In disease, the same mechanisms help alleviate problems created by malfunction of one or more of the body’s components. The remarkable complexity of these regulatory systems allow the body to meet a wide range of challenges, while numerous redundancies help avoid the consequences of Murphy’s law, which states: “If anything can go wrong, it will; and at the worst possible time.” Furthermore, damage to one or more of the mechanisms that maintain homeostasis often produces few ill effects, and sometimes none, because the body addresses Murphy’s law with another law: “If something is worth doing, many different mechanisms will see to it that this is done correctly.” The latter is well known to modern engineers, who design elaborate systems with redundancies that prevent failure of any component from causing the structure to crash.
Utilization of physiological regulatory mechanisms requires signals to inform a structure, such as the heart or a blood vessel, that its function must be modified, along with the means to initiate appropriate responses. To ensure that these responses are turned on when needed, operate at an appropriate intensity, and end when no longer useful, cardiovascular signaling systems collect and integrate information from many sources. Appropriate safeguards are provided by interlocking control mechanisms that recognize challenges, amplify signals, and adjust and integrate the responses that compensate for changing hemodynamics.
Two different types of response adjust cardiovascular function to the changing needs of the body (Fig. 8-1). The first are short-term functional responses that adjust the circulation to stresses—such as exercise and hemorrhage—that develop rapidly, over a few seconds or minutes, and generally last no longer than a few minutes or hours. The second are prolonged proliferative (transcriptional) responses that are evoked by chronic abnormalities, such as hypertension, a
leaky or stenotic heart valve, or after a myocardial infarction has irreversibly damaged the left ventricle. The latter, which evolve over periods of weeks, months, and often years, are mediated by changes in gene expression and protein synthesis that alter the architecture of the heart, and the size, shape, and composition of its myocardial cells.
leaky or stenotic heart valve, or after a myocardial infarction has irreversibly damaged the left ventricle. The latter, which evolve over periods of weeks, months, and often years, are mediated by changes in gene expression and protein synthesis that alter the architecture of the heart, and the size, shape, and composition of its myocardial cells.
 Fig. 8-1: Functional and proliferative responses. Functional responses, which alter the properties of preexisting structures by posttranslational mechanisms, aid survival by evoking responses like fight and flight. Proliferative responses, which take longer to evolve, allow organisms to grow out of trouble. (Images from Huber, 1981.) |
Functional Signal Transduction
Functional responses are often referred to as neurohumoral responses because most are evoked by the autonomic nervous system and chemical mediators that are secreted into the blood or extracellular fluid. The familiar cardiovascular response to exercise, which helps maintain arterial blood pressure and increase blood flow to the exercising muscles, is initiated largely by increased sympathetic activity and epinephrine released by the adrenal medulla. Along with decreased parasympathetic activity, these neurohumoral signals act on the heart and blood vessels to increase ventricular ejection and filling, accelerate heart rate, and constrict peripheral arterioles and veins. Norepinephrine, the most important sympathetic mediator, activates multistep signaling cascades that can be viewed as an old-fashioned bucket brigade (Fig. 8-2). However, unlike a bucket brigade, where a single substance (water) is passed from fireman to fireman, biological signaling cascades utilize many different signaling molecules and chemical reactions.
The first step in the response of the heart to sympathetic stimulation is norepinephrine release from sympathetic nerve endings into the extracellular space (Fig. 8-2). The second step, norepinephrine binding to β-receptors on the surface of cardiac myocytes, also takes place in the extracellular fluid, after which all of the remaining steps occur within these cells. These include G protein activation (step 3), which increases the catalytic activity of adenylyl cyclase (step 4), which generates cyclic AMP (step 5), which activates a cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA, step 6), which catalyzes the phosphorylation of L-type calcium channels (step 7), which increases the entry of “trigger” calcium into the cytosol (step 8), which increases the opening of intracellular calcium release channels (step 9), which releases more calcium for binding to troponin C (step 10), which increases the number of exposed actin molecules in the thin filaments (step 11), which increases the number of actin–myosin interactions
(step 12), which increases contractility (step 13), which increases the volume of blood ejected by the heart (step 14). Cascades similar to that in Figure 8-2, although different in detail, mediate virtually every signal that modifies cardiovascular function.
(step 12), which increases contractility (step 13), which increases the volume of blood ejected by the heart (step 14). Cascades similar to that in Figure 8-2, although different in detail, mediate virtually every signal that modifies cardiovascular function.
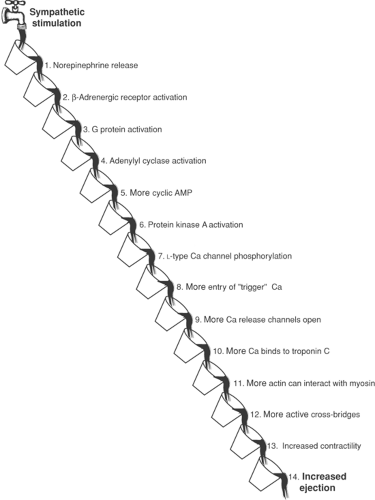 Fig. 8-2: Depiction of the signal cascade by which sympathetic stimulation increases ejection. This analogy shows 14 steps as a series of buckets where, in each step, an “upstream” signal causes the bucket to pour its contents into the next bucket, thereby transmitting the signal down the cascade. The response begins when sympathetic stimulation releases norepinephrine from nerve endings at the surface of cardiac myocytes (step 1), after which this neurotransmitter binds to and activates β-adrenergic receptors (step 2). The remaining steps, which all take place within myocardial cells, include activation of a G protein (step 3), increased adenylyl cyclase activity (step 4), accelerated production of cAMP (step 5), activation of a cAMP-dependent protein kinase (step 6), phosphorylation of plasma membrane L-type calcium channels (step 7), more calcium entry into the cell (step 8), increased opening of calcium release channels (step 9), increased calcium binding to troponin C (step 10), greater availability of actin in the thin filaments for interaction with myosin (step 11), participation of more cross-bridges in contraction (step 12), increased contractility (step 13), and a greater extent of ejection (step 14). |
Why are there so many Steps in a Signal Transduction Cascade?
One might ask why there are so many steps between a challenge, like exercise, and a physiological response like that depicted in Figure 8-2. The answer lies in the ability of biological signal transduction cascades, whose complexity might, at first glance, seem to be almost perverse, to enhance regulatory control. These cascades can amplify, inhibit, and fine-tune responses, integrate signal transduction cascades with one another, and prevent responses from going out of control (“runaway signaling”) by allowing signals to turn themselves off automatically. In fact, the depiction of the signal cascade in Figure 8-2 is oversimplified because most biological signaling pathways are not linear, where each step is coupled to a single downstream reaction, but instead form branches, loop forward and backward, interconnect with other pathways, and generate multiple signals at many steps. These intricacies organize responses that provide options for both amplification and negative feedback, and for the integration of each step in the response with other steps in the cascade and with additional signaling systems. This fine-tuning would not be possible if cell signaling generated only an “all-or-none” response.
An example of a mechanism that regulates signal transduction is the allosteric effect of ATP described in Chapter 7, which accelerates ion fluxes through channels, pumps, and exchangers. This effect is important in energy-starved hearts, where a fall in ATP concentration inhibits several steps in the signaling cascade shown in Figure 8-2; these include the opening of L-type calcium channels in the plasma membrane (step 8) and calcium release channels in the sarcoplasmic reticulum (step 9), and the interactions between actin and myosin (step 12). These and other allosteric effects of decreased ATP concentration help match the rates of ATP production and ATP consumption by inhibiting contraction, the most expensive energy-consuming reaction in the heart. Acidosis, which occurs when glycolysis is accelerated in energy-starved hearts (Chapter 2), also reduces energy consumption by inhibiting many steps in this cascade. A third example, which illustrates how a reaction near the “top” of a signaling pathway can respond to changes further down the cascade, occurs when increased calcium release into the cytosol (step 9) inhibits adenylyl cyclase (step 4); this negative feedback, by slowing the cascade, reduces contractility. Additional mechanisms that avoid calcium overload include the ability of increased cytosolic calcium to accelerate calcium transport out of the cytosol by the plasma membrane and sarcoplasmic reticulum calcium pumps, and by the sodium/calcium exchanger (Chapter 7).
Biological signal transduction cascades often allow a single stimulus to evoke an integrated, multifaceted response. For example, the elevated level of cAMP (step 5 in Fig. 8-2) increases the rate and extent of relaxation when phosphorylation of phospholamban accelerates calcium uptake into the sarcoplasmic reticulum (Chapter 7). Another example of signal diversification occurs at step 3 in Figure 8-2, where norepinephrine binding to a single β1-receptor activates both Gαs and Gβγ, each of which can modify a different downstream target (see below).
Control of cell function is enhanced further by a large number of isoforms of many signaling molecules. For example, binding of norepinephrine to β1– and β2-receptors in the heart evokes different responses, while in vascular smooth muscle, activation of α1-receptors by norepinephrine causes vasoconstriction, and β-receptor activation has a relaxing effect. These intricacies also
characterize intracellular signal transduction, as is evident by the ability of protein kinases to activate several pathways by phosphorylating a number of different signaling proteins.
characterize intracellular signal transduction, as is evident by the ability of protein kinases to activate several pathways by phosphorylating a number of different signaling proteins.
The Hemodynamic Defense Reaction
The hemodynamic defense reaction, which compensates for underfilling of the arterial system, is an integrated functional response that involves the heart, blood vessels, and kidneys (Table 8-1). As detailed in a brilliant essay by Poter Harris (1983), exercise, hemorrhage, and heart failure all initiate this response by stimulating cardiac performance, constricting blood vessels, and inhibiting salt and water excretion by the kidneys (Table 8-2). In exercise, a short-term challenge in which arterial blood pressure falls because of vasodilatation in the active muscles, the hemodynamic defense reaction restores blood pressure by stimulating the heart and constricting the arterioles supplying nonexercising tissues such as the gut and kidneys. Cardiac stimulation and selective vasoconstriction appear rapidly, over a few seconds or minutes, and last until the exercise ends, usually for a few minutes or hours. Urine output also decreases during exercise, but because the challenge is generally brief—marathon runners are a notable exception—fluid retention does not become apparent. More prolonged responses, supplemented by thirst and fluid retention by the kidneys, are initiated when cardiac output is severely decreased in a syndrome called shock. The latter can last for hours, at most a few days, after which the patient either dies or recovers. In heart failure, which is almost always progressive, the hemodynamic defense reaction persists and fluid retention commonly becomes a major clinical problem (see Chapter 18).
Exercise
The neurohumoral response evoked by exercise, the briefest of the challenges listed in Table 8-1, is initiated by several mechanisms. Most important is the baroreceptor reflex, which increases sympathetic activity and reduces parasympathetic tone in response to the fall in arterial blood pressure caused by vasodilatation in the exercising muscles. Sympathetic activity is also increased when CO2 and lactic acid released by the exercising muscles stimulate the chemoreceptors. These
responses are supplemented by nerve impulses that arise in the exercising muscles and the cerebral cortex; the latter explains the onset of sympathetic stimulation before the start of exercise, for example, when a sprinter hears the starter’s gun.
responses are supplemented by nerve impulses that arise in the exercising muscles and the cerebral cortex; the latter explains the onset of sympathetic stimulation before the start of exercise, for example, when a sprinter hears the starter’s gun.
Table 8-1 Three Conditions That Evoke the Hemodynamic Defense Reaction | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 8-2 The Hemodynamic Defense Reaction | |
|---|---|
|
The short-term neurohumoral response evoked by exercise is mediated by an inotropic effect that increases the amount of blood ejected during systole, a lusitropic effect that augments filling during diastole, and a chronotropic effect that accelerates heart rate (Tables 8-1 and 8-2). Together, these responses increase the flow of blood out of the heart (cardiac output). Sympathetic stimulation also causes contraction of vascular smooth muscle. Constriction of small arterioles helps maintain blood pressure by increasing the resistance in vascular beds not dilated by exercise, while venoconstriction increases the return of blood to the heart. Fluid retention, as noted above, becomes important only during prolonged exercise.
Shock
Shock, a clinical syndrome whose most obvious abnormalities are low blood pressure and inadequate tissue perfusion, also activates the hemodynamic defense reaction. There are many causes of this syndrome, which usually lasts no more than several hours (Table 8-3). Hypovolemic shock occurs when hemorrhage, diarrhea, or leaky capillaries severely decrease circulating blood volume. In distributive shock, which is caused by excessive vasodilatation, there is enough blood in the vasculature but it is in the wrong place, that is, the veins rather than the arteries. Distributive shock can be caused by vasodilator actions of endotoxins (e.g., toxic shock, gram-negative septicemia), reflex vasodilatation (vasovagal syncope, which is the old fashioned “swoon”), and the von Bezolt–Jarisch reflex which can be activated in patients with inferior or posterior myocardial infarction (Chapter 17). A third type of shock, cardiogenic shock, is seen after myocardial infarction, but in this case the cause is reduced ejection by a severely damaged left ventricle. Other causes of cardiogenic shock include valve rupture, pulmonary embolus, pericardial tamponade, acute myocarditis, and arrhythmias. If the underlying cause is reversible and appropriate therapy started promptly, patients usually
recover. However, if treatment is delayed this syndrome becomes irreversible because, even if blood pressure can be increased, the tissue damage initiated by low blood flow causes the patient to die.
recover. However, if treatment is delayed this syndrome becomes irreversible because, even if blood pressure can be increased, the tissue damage initiated by low blood flow causes the patient to die.
Table 8-3 Causes of Shock | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
The neurohumoral response in shock differs from that evoked by exercise because the challenge is more intense and, more importantly, lasts longer. The hemodynamic defense reaction, therefore, has time to cause the kidneys to retain salt and water, which facilitates restoration of blood volume. However, when vasoconstriction is prolonged and severe, organ damage by low flow is increased.
Patients can recover from shock if the underlying cause is reversible and appropriate treatment is started promptly. Otherwise, severe tissue damage causes this syndrome to become irreversible. If this occurs these patients are doomed to die even if blood pressure can be briefly restored.
Heart Failure
In heart failure, where underfilling of the arterial system usually lasts for a lifetime, most of the functional responses listed in Tables 8-1 and 8-2 become maladaptive. Tachycardia and increased contractility, which initially help maintain cardiac output, also increase cardiac energy utilization which, because most failing hearts are energy-starved, contributes to myocardial cell death. Arteriolar vasoconstriction helps maintain blood pressure when the heart fails, but the increased afterload reduces cardiac output and increases myocardial energy demand. Fluid retention by the kidneys, which is beneficial in patients with shock, represents a serious problem in heart failure because the elevated blood volume increases the already elevated pulmonary and systemic venous pressures to levels that cause fluid to be transuded into the lungs, peripheral tissues, and body cavities. Furthermore, the increased preload caused by venoconstriction and fluid retention can do little to increase cardiac output because Starling curves are often flattened in heart failure. For all of these reasons, the neurohumoral response usually becomes deleterious in most patients with heart failure (see Chapter 18).
Cardiac stimulation, vasoconstriction, and salt and water retention, which provide short-term support for the circulation during the brief challenges posed by exercise and shock,
become maladaptive in chronic heart failure. This paradox is vividly summarized by Harris (1983):
become maladaptive in chronic heart failure. This paradox is vividly summarized by Harris (1983):
Success and survival in the animal kingdom have overwhelmingly depended on physical mobility and strength. To ensure this the body makes use of the neuro-endocrine defense reaction which is also life-saving in injury … When the output of the [failing] heart decreases, the body reacts in the way nature has programmed it. It cannot distinguish. But now the neuro-endocrine response persists. Over weeks or months or years the retention of saline threatens the cardiac patient with drowning in his own juice. And every hour of every day he is running for his life.
Regulatory and Counterregulatory Responses
The hemodynamic defense reaction activates two opposing types of functional responses (Table 8-4). Regulatory responses, which include cardiac stimulation, vasoconstriction, and fluid retention help maintain blood pressure and cardiac output (see above). At the same time, however, counterregulatory responses that oppose the dominant regulatory responses are also evoked; these include a negative inotropic effect on the heart, vasodilatation, and diuresis. The ability of a single stimulus to activate opposing responses illustrates one way that biological signaling systems minimize the risk of “runaway signaling” (see below).
Table 8-4 Regulatory and Counterregulatory Neurohumoral Responses | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 8-5 Regulatory and Counterregulatory Neurohumoral Responses | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The functional responses evoked by some neurohumoral mediators are predominantly regulatory, whereas others are mainly counterregulatory. As a rule, regulatory mediators also have proliferative effects that promote cardiac hypertrophy, while counterregulatory mediators generally inhibit cell growth and proliferation (Table 8-5). However, there are exceptions to this generalization, and many neurohumoral mediators evoke both regulatory and counterregulatory responses.
Extracellular Signaling Molecules
The ability of chemical mediators to regulate cardiovascular function was discovered by Oliver and Scháfer (1895), who found that injection of adrenal extracts increases heart rate in anesthetized cats. A decade later, Elliott (1905) suggested that the response to sympathetic nerve stimulation, which resembles that evoked by adrenal extracts, might also be mediated by a chemical messenger. However, it was not until 1921 that Loewi (1921) carried out a simple but inspired experiment
which proved conclusively that a chemical can mediate a neurohumoral response, in this case, to vagal stimulation. Early efforts to isolate a chemical mediator had proven fruitless because the neurotransmitter is inactivated very rapidly, but Loewi overcame this problem by placing two frog hearts a short distance apart in a slowly moving stream of Ringer’s solution. When he stimulated the vagus nerve supplying the upstream heart, not only did this heart slow, but the rate of beating also decreased in the unstimulated downstream heart; in contrast, stimulation of the vagus nerve supplying the downstream heart had no effect on the upstream heart. This elegant experiment proved that vagal stimulation releases a chemical that slows the heart; a few years later Loewi, who initially called this chemical vagusstoff, identified the mediator as acetylcholine.
which proved conclusively that a chemical can mediate a neurohumoral response, in this case, to vagal stimulation. Early efforts to isolate a chemical mediator had proven fruitless because the neurotransmitter is inactivated very rapidly, but Loewi overcame this problem by placing two frog hearts a short distance apart in a slowly moving stream of Ringer’s solution. When he stimulated the vagus nerve supplying the upstream heart, not only did this heart slow, but the rate of beating also decreased in the unstimulated downstream heart; in contrast, stimulation of the vagus nerve supplying the downstream heart had no effect on the upstream heart. This elegant experiment proved that vagal stimulation releases a chemical that slows the heart; a few years later Loewi, who initially called this chemical vagusstoff, identified the mediator as acetylcholine.
In the 1930s, Cannon and Rosenblueth (1937) postulated that sympathetic stimulation releases a vasoconstrictor that they called sympathin E (for excitatory) which is related to epinephrine (adrenaline), a vasodilator that had previously been isolated from the adrenal medulla (which they called sympathin I, for inhibitory). Shortly after World War II, von Euler (1946) showed that the sympathetic neurotransmitter is norepinephrine, which differs from epinephrine only in the absence of a methyl group.
A variety of signaling molecules are known to regulate cardiovascular function (Table 8-6). These include tyrosine metabolites called catecholamines (e.g., epinephrine, norepinephrine, dopamine), quaternary amines (e.g., acetylcholine), other small organic molecules (e.g., thyroxin,
purines), peptides (e.g., angiotensin II, arginine vasopressin, natriuretic peptides, endothelin, cytokines, growth factors), steroid hormones (e.g., aldosterone), fatty acid derivatives (e.g., prostaglandins), and even a free radical gas (nitric oxide). Because these chemical mediators act on the outsides of cells, they are sometimes referred to as extracellular messengers.
purines), peptides (e.g., angiotensin II, arginine vasopressin, natriuretic peptides, endothelin, cytokines, growth factors), steroid hormones (e.g., aldosterone), fatty acid derivatives (e.g., prostaglandins), and even a free radical gas (nitric oxide). Because these chemical mediators act on the outsides of cells, they are sometimes referred to as extracellular messengers.
Table 8-6 Some Receptors and Extracellular Messengers That Modify Cardiovascular Function | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Interactions between Extracellular Messengers (Ligands) and their Receptors
Extracellular messengers are frequently called ligands because they bind with high affinity and specificity to receptors that recognize their presence as a signal to modify cell function. Most physiological ligands, as well as a majority of clinically useful drugs, are amphipathic molecules whose hydrophilic moieties prevent them from crossing the lipid barrier in biological membranes (Chapter 1). For this reason, their cellular actions depend on interactions with plasma membrane receptors. Exceptions include steroid and thyroid hormones, which are hydrophobic molecules that enter the cytosol where they interact with intracellular receptors.
The concept of specific receptors originated when Ahlquist (1948) found that low concentrations of epinephrine dilate blood vessels whereas higher concentrations cause vasoconstriction. This led him to postulate that epinephrine can bind to two types of receptors: α-receptors, which have a low affinity for this ligand and are responsible for the constrictor action seen at the higher epinephrine concentrations, and β-receptors which, because they have a higher affinity for epinephrine, mediate the vasodilator response seen at low concentrations of this extracellular messenger. The subsequent elucidation of the structures of these and other receptor molecules, along with the demonstration that different signal transduction pathways are activated when a given ligand interacts with different receptors, have proven Ahlquist’s hypothesis to be correct.
Signal Transmission
Endocrine (hormonal) signaling (Table 8-7) was discovered by Bayliss and Starling (1902), who observed that instilling acid into the duodenum stimulates pancreatic secretion, even after both tissues are denervated. After finding that intravenous injection of a jejunal mucosa extract also
stimulates pancreatic secretion, they proposed that a substance, which they called secretin, travels through the bloodstream from the jejunum, where it is produced, to the pancreas, where it evokes the response. Less than 20 years after this discovery, Loewi described neurotransmitter signaling, where the extracellular messenger is released by the nervous system (see above). More recently, signaling molecules were found to reach their receptors by shorter routes; in paracrine signaling an extracellular messenger released by one cell diffuses to a receptor on a nearby cell, while in autocrine signaling a cell modifies its own function by releasing a ligand that binds to a receptor on its surface. Cytoskeletal signaling allows mechanical interactions within and between neighboring cells and the surrounding extracellular matrix to alter cell structure and function. This type of signaling is initiated when cell deformation modifies signaling proteins—many of which are homologous to the ligands, receptors, enzymes, and other proteins that participate in cell signaling—that interact with the cytoskeleton (see Chapter 5).
stimulates pancreatic secretion, they proposed that a substance, which they called secretin, travels through the bloodstream from the jejunum, where it is produced, to the pancreas, where it evokes the response. Less than 20 years after this discovery, Loewi described neurotransmitter signaling, where the extracellular messenger is released by the nervous system (see above). More recently, signaling molecules were found to reach their receptors by shorter routes; in paracrine signaling an extracellular messenger released by one cell diffuses to a receptor on a nearby cell, while in autocrine signaling a cell modifies its own function by releasing a ligand that binds to a receptor on its surface. Cytoskeletal signaling allows mechanical interactions within and between neighboring cells and the surrounding extracellular matrix to alter cell structure and function. This type of signaling is initiated when cell deformation modifies signaling proteins—many of which are homologous to the ligands, receptors, enzymes, and other proteins that participate in cell signaling—that interact with the cytoskeleton (see Chapter 5).
Table 8-7 Routes by Which Extracellular Messengers Reach Cells | |||||
|---|---|---|---|---|---|
|
Most extracellular messengers contain hydrophobic moieties, so that these molecules have high partition coefficients, a variable that quantifies the distribution of a molecule between the lipid bilayer and the surrounding aqueous medium. Molecules with high partition coefficients tend to accumulate in membranes; for example, when a cell in an aqueous medium encounters 10,001 molecules with a partition coefficient of 10,000, 10,000 of the molecules will enter the bilayer, leaving only 1 in the aqueous medium. Because of their hydrophobicity, extracellular messengers and drugs often have partition coefficients in the thousands. The anti-arrhythmic drug amiodarone has a partition coefficient >1,000,000 (Herbette et al., 1988) that explains its remarkably long biological half-life, which is many months. Their high hydrophobicity allows these molecules to gain access to plasma membrane receptors by first dissolving in the lipid bilayer and then diffusing to their receptors (Herbette et al., 1991). Utilization of this lipid pathway, which allows ligands to gain rapid access to their membrane receptors, also explains why the ligand- and drug-binding sites of many intrinsic membrane proteins include hydrophobic regions of the membrane-spanning α-helices within the bilayer.
LigandBinding to Receptors: Receptor Number and Binding Affinity
The interactions between ligands and their receptors can be characterized in terms of the number of receptors that can bind to the ligand and the affinity with which the ligand attaches to the receptor. Receptor number is often quantified as Bmax, the maximal amount of ligand that binds specifically to the receptor. A commonly used index of the affinity of a receptor for its ligand is the dissociation constant (kd), which is the ligand concentration at which 50% of receptors are bound to the ligand; this means that the greater the affinity of the receptor for the ligand, the lower will be the kd. The constant kd also describes the ratio between the rate of ligand dissociation from its receptor (the “off-rate”) and the rate of binding of the ligand (the “on-rate”). Affinity can also be expressed as an association (binding) constant (kb), which is the reciprocal of kd, the dissociation constant. Ligands have a wide range of receptor-binding affinities; some peptides bind to their receptors at concentrations below 1012 M, while most neurohumoral transmitters and drugs occupy their receptors at concentrations between 108 and 106 M. A few compounds, like ethanol, interact with membrane proteins at much higher concentrations (Table 8-8).
Binding affinity is an important determinant of the specificity of the biological response to a ligand. A ligand that binds to its receptor with high affinity usually evokes a specific response
that is accompanied by few side effects; this is because low concentrations of the ligand can be recognized by the receptor, which minimizes interactions of the ligand with other cell components. Ligands that bind to their receptors with low affinity, and so evoke responses only at high concentrations, often have additional “nonspecific” actions that are usually undesirable. The toxic effects of most clinically useful drugs are seen at concentrations higher than those that produce the desired therapeutic effects; exceptions include allergic or sensitivity reactions, which are generally much less dependent on the concentration of the ligand. Specificity can be described as the ratio of the ligand concentrations that cause toxic and therapeutic effects; a higher “toxic/therapeutic ratio” means that a drug is less likely to cause unwanted side effects when administered at doses that yield desirable therapeutic effects.
that is accompanied by few side effects; this is because low concentrations of the ligand can be recognized by the receptor, which minimizes interactions of the ligand with other cell components. Ligands that bind to their receptors with low affinity, and so evoke responses only at high concentrations, often have additional “nonspecific” actions that are usually undesirable. The toxic effects of most clinically useful drugs are seen at concentrations higher than those that produce the desired therapeutic effects; exceptions include allergic or sensitivity reactions, which are generally much less dependent on the concentration of the ligand. Specificity can be described as the ratio of the ligand concentrations that cause toxic and therapeutic effects; a higher “toxic/therapeutic ratio” means that a drug is less likely to cause unwanted side effects when administered at doses that yield desirable therapeutic effects.
Table 8-8 Ligand-Binding Affinities of Some Cardiac Plasma Membrane Receptors | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Receptor Blockade
Characterization of specific receptors made it possible to identify drugs that could inhibit their ability to interact with their ligands. Such inhibitors, often called blockers or antagonists, usually have structures similar to those of the physiological extracellular messengers. The inhibitory effects of many of these drugs exhibit competitive kinetics and can be reversed by high concentrations of the physiological ligand. A few drugs inactivate a signal transduction system completely and permanently; aspirin, for example, acetylates and irreversibly inactivates cyclooxygenase, an enzyme that releases a thrombogenic prostaglandin (see below).
The difference between most receptor blockers (antagonists) and the physiological ligands (agonists) is not where these molecules bind, but what happens after binding has occurred. Unlike an agonist, which activates the subsequent steps in a signal transduction cascade (e.g., after step 1 in Fig. 8-2), antagonists occupy the receptor but do not generate an intracellular signal. The clinical value of an antagonist (e.g., a β-adrenergic blocker) therefore reflects the fact that the receptor-bound antagonist inhibits the ability of the receptor to bind to, and thus become activated by, the physiological agonist (e.g., norepinephrine).
Not all molecules that interact with receptors can be classified simply as agonists and antagonists. Some drugs, called partial agonists, bind to a receptor where they cause a weak activation of its signal transduction cascade, while at the same time blocking the ability of the receptor to interact with the more potent physiological agonists. In the case of the β-adrenergic blockers, the weak stimulatory effect of a partial agonist is referred to as intrinsic sympathomimetic activity (ISA). By blocking the more potent effects of norepinephrine, partial β-adrenergic agonists inhibit the response to surges of sympathetic activity while providing a low level of adrenergic stimulation in the basal state. Unfortunately, many partial agonists have turned out to be less advantageous clinically than this description might suggest.
Types of Receptors
Cardiovascular function is regulated when a variety of ligands bind to their receptors (Table 8-6). Most functional signals are mediated by G protein-coupled receptors (GPCRs), which are named for the heterotrimeric guanosine triphosphate (GTP)-binding proteins that mediate
their cellular actions (see below). Enzyme-linked receptors, most of which contain a kinase or other enzyme that is activated when the receptor binds to its ligand, were initially thought to mediate only proliferative responses (see Chapter 9); however, these receptors are now known to participate in functional signaling as well. Cytokine receptors resemble enzyme-linked receptors except that instead of having intrinsic enzyme activity, ligand-bound cytokine receptors modify the catalytic activity of other enzymes. Ion channel-linked receptors contain channels that are opened when the receptor binds its ligand. Nuclear receptors bind to hormones, like aldosterone and thyroxin, whose hydrophobic structure allows them to cross the plasma and nuclear membranes; the resulting ligand-receptor complexes regulate gene expression by interactions with DNA in the nucleus. Cellular responses to nitric oxide are not mediated by a receptor; instead, this signaling molecule binds directly to guanylyl cyclase, its target within cells.
their cellular actions (see below). Enzyme-linked receptors, most of which contain a kinase or other enzyme that is activated when the receptor binds to its ligand, were initially thought to mediate only proliferative responses (see Chapter 9); however, these receptors are now known to participate in functional signaling as well. Cytokine receptors resemble enzyme-linked receptors except that instead of having intrinsic enzyme activity, ligand-bound cytokine receptors modify the catalytic activity of other enzymes. Ion channel-linked receptors contain channels that are opened when the receptor binds its ligand. Nuclear receptors bind to hormones, like aldosterone and thyroxin, whose hydrophobic structure allows them to cross the plasma and nuclear membranes; the resulting ligand-receptor complexes regulate gene expression by interactions with DNA in the nucleus. Cellular responses to nitric oxide are not mediated by a receptor; instead, this signaling molecule binds directly to guanylyl cyclase, its target within cells.
Table 8-9 Some Important G Protein-Coupled Receptors in the Cardiovascular System | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


