Fig. 4.1
Example of increased left stellate ganglion nerve activity (SGNA) preceding ventricular fibrillation (VF) and sudden cardiac death (Zhou et al. 2008). (a) Increased low-amplitude burst discharge activity (LABDA) resulted in accelerated idioventricular rhythm. (b) VF occurred approximately 40 s later. Panels (a) and (b) are continuous. (c) A 6-s recording from panel (b). INA integrated nerve activity in millivolts, P P wave, which is dissociated from ventricular activation due to complete AV block (Reprinted with permission from Elsevier)
4.3 Role of Adrenergic Receptor Activation
Enhanced sympathetic nerve activity increases vulnerability to ventricular arrhythmias in the ischemic heart by complex processes. Multifold direct arrhythmogenic effects on cardiac electrophysiologic function are primarily mediated through β1-adrenergic receptors. They include derangements in impulse formation, conduction, repolarization alternans, and heterogeneity of repolarization, with the potential for culmination in ventricular tachycardia and fibrillation (Fig. 4.2) (Opie 2004; Lee et al. 1988). Indirect effects include impairment of oxygen supply–demand ratio resulting from increased cardiac metabolic activity, alpha-adrenergically mediated coronary vasoconstriction, especially in vessels with damaged endothelium, and changes in preload and afterload.
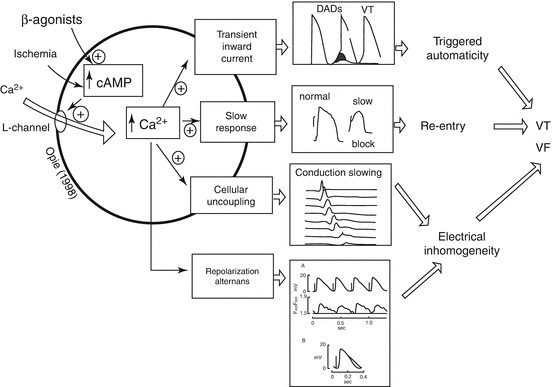
Fig. 4.2
The cardiac β-adrenergic signaling system mediating ventricular arrhythmogenesis. The central pathways include links between cyclic adenosine 3′,5′-monophosphate (cAMP), cytosolic calcium, and specific calcium-mediated electrophysiologic abnormalities that predispose to ventricular tachycardia (VT) and ventricular fibrillation (VF) (Modified from Opie (2004) and used with his permission). The lowest panel is based on Lee et al. (1988), indicating that simulated ischemia results in alternation in calcium transients, which appears to underlie action potential alternans (Reprinted with permission from Lippincott Williams & Wilkins)
Increased levels of catecholamines stimulate β-adrenergic receptors, which in turn alter adenylate cyclase activity and intracellular calcium flux. These effects are probably mediated by the cyclic nucleotide and protein kinase regulatory cascade, which can alter spatial heterogeneity of calcium transients and consequently provoke T-wave alternans and heterogeneity of repolarization (Verrier et al. 2011).
In the setting of myocardial ischemia, α-adrenergic blockade may alleviate coronary vasoconstriction and reduce platelet aggregability, but in the normal heart, α-adrenergic receptor stimulation or blockade does not appear to affect ventricular electrical stability, as evidenced by the fact that administration of an α-adrenergic agonist such as phenylephrine or methoxamine does not influence excitable properties when the pressor response is controlled to prevent reflex changes in autonomic tone (Verrier et al. 1974; Kowey et al. 1983).
4.4 Sympathetic–Parasympathetic Nerve Interactions
Vagus nerve influences are contingent on the prevailing level of adrenergic tone (Lown and Verrier 1976). When sympathetic nerve activity is augmented by thoracotomy, sympathetic nerve stimulation, myocardial ischemia, or catecholamine infusion, vagus nerve activation exerts a protective effect on ventricular vulnerability. But, vagus nerve stimulation alone is without effect on ventricular vulnerability during β-adrenergic blockade. Levy and Blattberg (1976) termed this phenomenon “accentuated antagonism” (Fig. 4.3) (Opie 2004). The basis for this antagonism of adrenergic effects is presynaptic inhibition of norepinephrine release from nerve endings and muscarinically mediated action at the second messenger level, attenuating the response to catecholamines at receptor sites (Levy and Blattberg 1976). Vagus nerve influences also provide indirect protection against ventricular fibrillation during both myocardial ischemia and reperfusion by reducing excess heart rates, which can otherwise critically compromise diastolic perfusion time to increase the ischemic insult (Zuanetti et al. 1987). However, the beneficial effects of vagus nerve activity may be annulled if profound bradycardia and hypotension ensue.
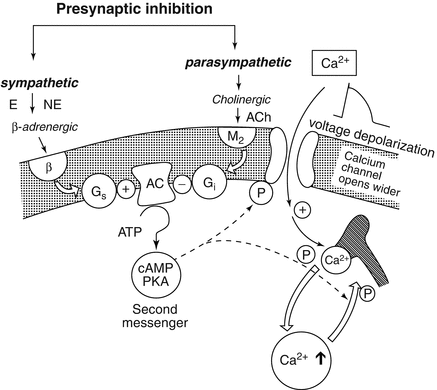
Fig. 4.3
Interaction between parasympathetic and sympathetic systems at a cellular level may involve two opposing cyclic nucleotides, cyclic adenosine 3′,5′-monophosphate (cAMP) and cyclic guanosine 3′,5′-monophosphate. Many effects of vagal stimulation can best be explained by the inhibitory effect on the formation of cAMP, including formation of the inhibitory G protein Gi in response to M2-receptor stimulation (Reprinted from Opie (2004) and used with his permission)
4.5 Nerve Degeneration and Regrowth in Response to Myocardial Infarction
Myocardial infarction can elicit extensive damage to the afferent and efferent innervation of the heart (Minardo et al. 1988) (Fig. 4.4). The resulting heterogeneous reinnervation, supersensitivity to catecholamines, and loss of antiarrhythmic effects of vagus nerve activity converge to enhance susceptibility to ventricular arrhythmias during the acute phase of myocardial infarction (Table 4.1) (Zipes and Miyazaki 1990).
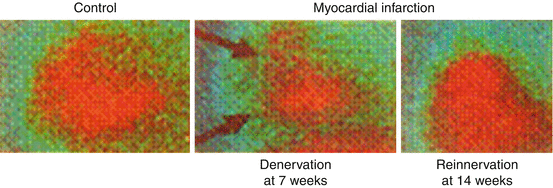
Fig. 4.4
MIBG scintigraphic images of sympathetic innervation before and after myocardial infarction in dogs (Minardo et al. 1988). Left panel: Left lateral preoperative metaiodobenzylguanidine (MIBG) image showing homogeneous uptake. Middle panel: MIBG images obtained 7 weeks after latex injection showing anteroapical defect (arrow). Right panel: MIBG images at 14 weeks after latex injection showing homogeneous uptake (Reprinted with permission from Lippincott Williams & Wilkins)
Table 4.1
Autonomic effects of myocardial infarction
Hours: Damage to efferent and afferent autonomic nerve supply |
Days, weeks: |
Heterogeneous re-innervation (14 weeks) |
Denervation supersensitivity (>17 weeks) |
Risk for arrhythmias |
Desensitization to cardiac pain, due to damage to afferent fibers |
Chen and coworkers (2001) demonstrated a significant correlation between increased sympathetic nerve density as reflected in immunocytochemical markers and history of ischemia in native hearts of human transplant recipients. In a canine model, they documented increased incidence of ventricular tachycardias and sudden cardiac death following induction of nerve sprouting with nerve growth factor. Episodes of T-wave alternans also occurred (Tsai et al. 2002), consistent with this parameter’s capacity to track arrhythmia vulnerability in humans as well as animal models (Verrier et al. 2011). The predisposition to arrhythmias was also linked to immunocytochemical evidence of a heterogeneous pattern of sympathetic nerve reinnervation (Zhou et al. 2004; Verrier and Kwaku 2004) (Fig. 4.5), prompting the suggestion that the term “neural remodeling” should be employed alongside “myocardial remodeling” in the conceptual framework of the pathophysiology of acute myocardial infarction.
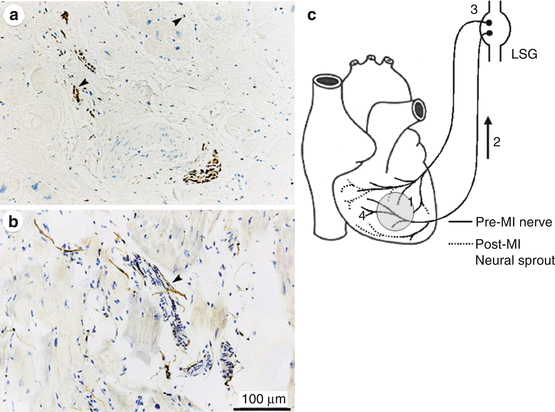
Fig. 4.5




Nerve sprouting after myocardial infarction. Panels (a) and (b) demonstrate TH-positive nerve fibers (arrowheads) in injured areas or around coronary arteries and in a patient with coronary artery disease (Cao et al. 2000). Panel (c) signaling of neural remodeling after myocardial infarction (Verrier and Kwaku 2004). Myocardial injury (shaded area) results in early local nerve growth factor (NGF) release, presumably from damaged cells, followed by upregulated NGF and growth-associated protein 43 (GAP43) expression, especially in the infarct area (1). These signal proteins are then retrogradely transported (2) to the nerve cell bodies in the ganglia (3) where they stimulate the sprouting of new cardiac nerve endings in the heart (4), predominantly in noninfarcted regions, leading to heterogeneous hyperinnervation (Reprinted with permission from Lippincott Williams & Wilkins)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
