Justin C. Mason Inflammatory rheumatic diseases have a long recognized relationship with the cardiovascular system. Because the treatment of many rheumatic diseases has improved considerably over the last 15 years and increased survival, the importance and complexity of this interrelationship have achieved prominence. Patients with multisystem rheumatic diseases may on occasion initially be evaluated by a cardiovascular specialist, a cardiologist, or a vascular or cardiothoracic surgeon, and early recognition of the immune-mediated basis of the cardiovascular disease reduces morbidity and mortality. The vasculature may represent a primary target organ of the underlying rheumatic disease and can be affected at numerous sites and at all levels. Thus large-vessel vasculitides may affect the entire aortic wall. Systemic sclerosis (SSc) commonly results in pulmonary arterial vasculopathy and pulmonary artery hypertension (PAH). Antineutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitides (AASVs) affect arterioles preferentially. Antiphospholipid syndrome (APS) causes both venous and arterial thromboses. Cardiac complications of systemic lupus erythematosus (SLE) include coronary arteritis, pericarditis, myocarditis, and valvular heart disease. Renal artery stenosis leading to uncontrolled hypertension is a feature of Takayasu arteritis (TA), and occlusive lesions in the subclavian, axillary, or iliac arteries may lead to limb claudication in patients with TA and giant cell arteritis (GCA). Rheumatic diseases have equally important secondary effects on the cardiovascular system. Chronic systemic inflammation predisposes to endothelial dysfunction and increased arterial stiffness, thereby escalating risk for the development of atherosclerosis. Cardiovascular specialists are increasingly recognizing the significantly increased prevalence of premature myocardial infarction and stroke in patients suffering from rheumatoid arthritis (RA) and SLE. Many outstanding clinical challenges remain, and predominant among them are early recognition and diagnosis of patients with rheumatic disease who have the highest risk for cardiovascular complications, alongside improved understanding of the underlying molecular mechanisms and the development of preventive strategies. Recognition of the role of inflammation in atherosclerosis has highlighted and stimulated study of the potential relationship between systemic inflammatory diseases and accelerated atherogenesis. This effort has substantially advanced our understanding of both the underlying pathogenic mechanisms and epidemiology. Current priorities include identification of patients most at risk and the development of preventive therapeutic strategies.1 Evidence supporting an association between inflammatory diseases and accelerated atherogenesis is best developed for RA and SLE. In addition, ankylosing spondylitis, psoriatic arthritis, AASV, TA, and APS may all be associated with premature atherosclerosis. Cardiovascular specialists should consider an underlying inflammatory disease in young patients with otherwise unexplained angina, myocardial infarction, or stroke. Patients with a rheumatic disease who suffer a myocardial infarction have worse outcomes in terms of both heart failure and mortality than does the age-matched general population.2 Homeostatic mechanisms promote a quiescent, antithrombotic, antiadhesive vascular endothelium and control vasodilation and permeability (see Chapters 41 and 49). Prolonged systemic inflammation such as that seen in RA and SLE may promote endothelial injury, increased endothelial apoptosis, and endothelial vasodilator dysfunction. Traditional risk factors alone do not explain the increased burden of atherosclerosis, but inflammation may exacerbate the effects of classic risk factors.3 When compared with the general population, patients with systemic inflammatory diseases more commonly exhibit endothelial dysfunction and increased aortic stiffness. Although the results of individual studies vary, effective treatment of the underlying inflammation may not always reverse the endothelial dysfunction or improve the aortic stiffness.4,5 This observation has led to the hypothesis that the systemic inflammatory environment may predispose to increased plaque instability and rupture, a conjecture supported by an autopsy study.6 Given the increased plaque burden reported, both accelerated atherogenesis and higher-risk plaque may contribute to the observed increased incidence of premature cardiovascular events. Various molecular mechanisms mediate the increased risk for atherosclerotic disease and cardiovascular events. In addition to the traditional cardiovascular risk factors, disease-related factors may include effects of the proinflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), and IL-6 on endothelial activation, leukocyte adhesion, endothelial injury, and permeability. Increased endothelial cell apoptosis and diminished capacity for repair may contribute. Autoantibodies (e.g., antiphospholipid antibodies), CD4+CD28− cytotoxic T cells, Th17/TREG imbalance, complement deficiency or excessive activation, genetic polymorphisms, and the deleterious effects of drugs, including corticosteroids and cyclosporine, may also contribute.2,7 RA, an autoimmune, symmetric inflammatory polyarthritis with a female-to-male ratio of 3:1, affects up to 1% of the population in the Western world, with the onset of symptoms most commonly occurring between 30 and 50 years of age. Up to 80% of patients have a positive serum rheumatoid factor and/or anti–cyclic citrullinated peptide (CCP) antibody test. A systemic inflammatory response is evident, with low-grade fever, weight loss, raised erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), hypoalbuminemia, normochromic normocytic anemia, and thrombocytosis. A variety of studies have shown subclinical arterial disease with increased carotid intimal-medial thickness (IMT) and early plaque development. Although RA independently raises the risk for atherosclerosis, the precise mechanistic relationship between RA and atherogenesis remains unknown. Similarly, the mechanisms and long-term outcomes of abnormalities in myocardial perfusion and coronary flow reserve that have been reported in patients with RA and normal epicardial arteries remain to be established.8 A recent study of microvascular and macrovascular function in RA has suggested that the classic cardiovascular risk factors may influence endothelial function more than disease-related inflammation does.9 The initial abnormalities in vascular function may occur at or before the onset of RA symptoms. The direct effect of chronic inflammation on vascular endothelium may itself promote atherogenesis, in addition to exacerbating the actions of traditional cardiovascular risk factors. Moreover, the systemic inflammatory environment can contribute to the features of plaque and blood that promote cardiovascular events in patients with RA. Patients with RA have increased classic risk factors for atherosclerosis. Tobacco smoking is associated with both cardiovascular risk and the development of RA. Similarly, insulin resistance and the metabolic syndrome are more common in RA. Patients with RA may have a particular dyslipidemic pattern that includes high triglyceride levels and low levels of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol.7 The risk for myocardial infarction in patients with RA is similar to that in those with diabetes mellitus,10 and women with RA are twice as likely as age-matched controls in the general population to suffer myocardial infarction. Although death rates from both heart attack and stroke are comparable to that in the general population, events occur at an earlier age, with 50% of premature deaths in patients with RA being a direct consequence of cardiovascular disease. The excess mortality becomes apparent 7 to 10 years after diagnosis and has been associated with persistent disease activity and the presence of rheumatoid factor and anti-CCP antibodies. Current evidence suggests that patients with RA who suffer a myocardial infarction are less likely to receive acute reperfusion therapy and secondary preventive measures and thus have worse outcomes.11 Drug therapy for RA has undergone a remarkable evolution over the past 15 years, with a current focus on biologic therapies and aggressive management of early disease. Clinical trials have demonstrated that this approach reduces symptoms and structural damage to joints. It remains uncertain, however, whether drugs that control synovitis also confer vascular protection. Methotrexate is now the most widely used disease-modifying antirheumatic drug (DMARD), and since its introduction, mortality from myocardial infarction in patients with RA has improved. Similar observations have been made for sulfasalazine and hydroxychloroquine. Patients who do not respond adequately to DMARD therapy should switch to biologic therapies. Such agents now include those targeting TNF-α (infliximab, adalimumab, etanercept, certolizumab, and golimumab), the IL-6 receptor (tocilizumab), CTLA4Ig (abatacept), and the B cell–depleting monoclonal antibody rituximab. An aggressive disease-modifying approach also minimizes the use of nonsteroidal anti-inflammatory drugs (NSAIDs) and the requirement for corticosteroid therapy. Glucocorticoids may adversely affect traditional risk factors such as insulin resistance, hypertension, and lipid profiles and may hasten carotid plaque formation in RA. Because NSAIDs and cyclooxygenase-2 (COX-2)-selective NSAIDs (coxibs), although effective, may elevate blood pressure and increase the frequency of thrombotic cardiovascular events, caution is required regarding their use in patients with cardiovascular complications of inflammatory disease. However, evidence suggests that NSAID use in patients with RA does not confer an increased risk for cardiovascular events, thus indicating that their anti-inflammatory effects predominate.12 Demonstration of the potential cardiovascular benefits of the biologic therapies will require the results of long-term prospective studies (see later). TNF-α promotes vascular endothelial activation and dysfunction and may lead to plaque destabilization, and hence blockade would appear to be an attractive therapeutic option. Infliximab therapy may improve endothelial function as measured by flow-mediated dilation 4 to 12 weeks after infusion, whereas etanercept has been reported to reduce aortic stiffness. In contrast, a recent study found no change in macrovascular function in response to TNF-α blockade at 3 months,9 and infliximab therapy for 3 years had no effect on carotid IMT in patients with RA in comparison to controls. Data from the British Society of Rheumatology Biologics Register are more encouraging. Despite no reduction in the incidence of myocardial infarction in patients treated with a TNF-α antagonist as opposed to conventional DMARDs (methotrexate, sulfasalazine, and hydroxychloroquine), infarction was markedly reduced in anti–TNF-α responders versus nonresponders.13 Thus tight control of RA disease activity per se appears to have a beneficial effect on the risk for myocardial infarction. Treatment of the arthritis must be combined with a careful review of classic risk factors and appropriate steps taken to modify these factors. Although precise guidelines are awaited, most rheumatologists have a low threshold for addition of a statin, with the goal being a target LDL cholesterol level lower than 3.37 mmol/liter and HDL cholesterol higher than 1.04 mmol/liter.7 SLE, a systemic autoimmune disease, predominates in women at a ratio of 9:1 and affects all racial groups but more commonly those of Afro-Caribbean, Asian, and Chinese extraction. The reported prevalence varies from 4 to 280 per 100,000 population. Constitutional symptoms at initial evaluation include night sweats, lethargy, malaise, and weight loss. Frequent mucocutaneous features include the classic butterfly facial rash, oral ulcers, and alopecia. Serositis, myalgia, arthralgia, and Jaccoud nonerosive arthropathy also occur. Potentially life-threatening complications include glomerulonephritis leading to renal failure, central nervous system (CNS) involvement with cerebral vasculitis, pneumonitis, shrinking lung syndrome, and PAH. Hematologic involvement includes lymphopenia in most and frequently hemolytic anemia, neutropenia, and thrombocytopenia. Cardiac manifestations of SLE are relatively rare but include pericarditis, myocarditis, endocarditis, aortitis, and coronary arteritis. Understanding of the pathogenesis of SLE has improved significantly in recent years. A defect in the clearance of apoptotic cells results in the exposure of nuclear antigens to an immune system with hyperreactive B cells. Loss of immune tolerance results in the generation of autoantibodies and immune complexes. Deposition of immune complexes in target organs leads to activation of complement and tissue injury.14 Most patients have high-titer antinuclear antibodies and antibodies against double-stranded DNA (dsDNA). The latter are more specific for the diagnosis of SLE, and this is reinforced by the presence of antibodies against one or more nuclear antigens, including Sm, Ro, La, and ribonucleoprotein (RNP). Complement activation and consumption of C3 and C4 leading to reduced plasma levels characterize active disease. The ESR also rises in active disease, but CRP levels typically remain normal except in those with serositis or secondary infection. A bimodal peak in SLE-related mortality was first reported in the 1970s. The early peak was associated predominantly with active SLE and infectious complications secondary to immunosuppressive therapy, whereas coronary artery disease caused most deaths in the second peak. Since then, a variety of studies have suggested an increased risk for myocardial infarction and stroke in patients with SLE that is between 2-fold and 10-fold and up to 50-fold greater than that in the general population. The young age of patients with SLE and cardiovascular disease (67% of female patients with SLE and a first cardiac event are typically initially seen before 55 years of age) suggests that SLE accelerates arterial disease. In an inception cohort of 1249 patients monitored for 8 years, 97 vascular events were recorded, 31 of which resulted from atherosclerotic disease.15 Although the pattern of coronary artery disease in SLE does not appear to differ (Fig. 84-1), the plaque may be more vulnerable to rupture. Patients with SLE have worse outcomes following myocardial infarction than the age-matched general population does, with a higher risk for the development of cardiac failure and increased mortality.2 This difference may result from late diagnosis of ischemic heart disease and a reluctance to treat aggressively. Hypertension is common in SLE because of the presence of renal disease and the use of glucocorticoids in many patients. Similarly, patients with SLE commonly have metabolic syndrome, which is associated with renal impairment, higher corticosteroid doses, and Korean or Hispanic ethnicity.16 Patients with SLE also have lipid abnormalities, including high levels of very low-density lipoprotein (VLDL) and triglycerides, elevated or normal LDL cholesterol, and reduced HDL cholesterol. Moreover, proinflammatory HDL leading to increased oxidatively modified LDL cholesterol was seen in 45% of patients with SLE as compared with 20% of those with RA and 4% of the general population.2,7 Antibodies against oxidized LDL also occur in SLE and may promote atherogenesis. Mild SLE with rash and arthralgia can be treated with simple analgesics and NSAIDs, with the addition of hydroxychloroquine if required. Mild to moderate organ involvement, including mild renal impairment, hematologic abnormalities, myositis, arthritis, and cutaneous lesions, requires the addition of prednisone and typically an immunosuppressant such as azathioprine, mycophenolate mofetil (MMF), or methotrexate to aid in controlling the disease and allow steroid-sparing therapy. Cyclophosphamide and high-dose corticosteroids remain the first-line treatment of life-threatening complications, including myocarditis, cerebritis, severe hematologic involvement, and glomerulonephritis. MMF is increasingly being used in place of cyclophosphamide for lupus nephritis because of its equivalent efficacy and concerns regarding the risk for permanent infertility seen in up to 50% of patients treated with cyclophosphamide. Most rheumatologists consider rituximab an effective treatment of severe SLE, although clinical trials to date have proved disappointing, which may reflect the high doses of corticosteroids used in these trials and consequent masking of the benefits associated with rituximab. A variety of regimens have been used, including combinations of rituximab, prednisone, and cyclophosphamide. Belimumab, a monoclonal antibody that binds to the soluble B lymphocyte stimulator and prevents its interaction with B cell surface receptors, has a modest disease-modifying effect in patients with moderate non–renal-related SLE. Defining effective strategies for prevention of cardiovascular disease in patients with SLE will require long-term prospective trials with adjudicated cardiovascular endpoints. Undertreated and/or persistently active disease has been associated with accelerated atherogenesis. Therefore adequate individualized immunosuppressive therapy should minimize cardiovascular complications. Hydroxychloroquine reduces LDL cholesterol and lowers mortality from cardiovascular disease in patients with SLE. Aggressive management of traditional risk factors is also advocated, including diligent monitoring and tight blood pressure control. Statins are widely used, particularly in patients with renal impairment. Caution should be exercised in patients with active myositis, however, because statin therapy can exacerbate this complication. The clinical data available do not support significant protection against atherosclerosis by statins 2 to 3 years after initiation, although longer-term analysis is awaited.14 The relationship between chronic inflammation and atherogenesis implies that many rheumatic diseases may be associated with premature and increased cardiovascular risk (Table 84-1). Because data in support of this hypothesis are derived from relatively small studies, important current clinical challenges include the need to determine (1) which rheumatic diseases pose the greatest cardiovascular threat, (2) a means of identifying subsets of patients most at risk, and (3) preventative strategies to minimize cardiovascular events. TABLE 84-1 Coronary Artery Involvement and the Rheumatic Diseases Ankylosing spondylitis, psoriatic arthritis, and gout may also be associated with atherosclerotic disease. Hyperuricemia independently predicts cardiovascular disease, and patients with gout often have hypertension, hyperlipidemia, obesity, and diabetes mellitus. Many drugs used for the treatment of cardiac disease, including diuretics, beta blockers, and low-dose aspirin, can increase serum uric acid levels. In contrast, losartan, angiotensin-converting enzyme (ACE) inhibitors, atorvastatin, and fenofibrate may reduce urate levels.17 Allopurinol may reduce the risk for congestive cardiac failure and cardiovascular-associated death. In addition to achieving a serum uric acid level lower than 0.36 mmol/liter, patients with gout should receive dietary advice and aggressive management of cardiovascular risk factors. A systematic review of articles on cardiovascular disease in patients with psoriatic arthritis has revealed increased traditional risk factors, endothelial dysfunction, aortic stiffness, and subclinical atherosclerosis. The limited data available also suggest that adequate suppression of inflammatory disease activity leads to improvement in endothelial dysfunction and carotid IMT.18 Patients with ankylosing spondylitis have demonstrated impaired endothelial function and increased carotid IMT and pulse wave velocity, all of which indicate an increased risk for atherosclerosis.19 The impact of the increasing use of anti–TNF-α therapies on the incidence of cardiovascular events in these patients should emerge from international biologic registries. The vasculitides, a heterogeneous group of diseases, represent a significant clinical challenge, both diagnostically and therapeutically. The primary systemic vasculitides are classified into large-, medium-, and small-vessel disease. This leaves a small group of unclassified conditions, including Behçet disease, relapsing polychondritis, primary CNS vasculitis, and Cogan syndrome.20 The histologic features of vasculitis include perivascular inflammatory infiltrates that may invade the arterial wall, fibrinoid necrosis, thrombosis, fibrosis, and scar formation. Fibrinoid necrosis, a specific feature of the medium- and small-vessel vasculitides, typically affects the inner layer of the tunica media. Complications include stenosis and occlusions resulting in organ ischemia, thrombosis, aneurysm formation, and hemorrhage. Although biopsy is optimal for making the diagnosis, suitable tissue may not always be accessible, or arterial biopsy may be considered too hazardous, such as in patients with TA. Thus diagnosis often depends on clinical findings, laboratory indices, and imaging. The immunopathogenesis of the vasculitides is complex, multifactorial, and poorly understood. The endothelium may be subject to complement-mediated injury as a consequence of immune complex deposition in polyarteritis nodosa (PAN) or rheumatoid vasculitis. In the medium- and small-vessel vasculitides, ANCAs may activate neutrophils and subsequently damage the endothelium. The proinflammatory cytokines TNF-α, IL-1, IL-6, and interferon-gamma (IFN-γ) may activate the endothelium and induce the expression of adhesion molecules, including E-selectin, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1), thereby facilitating leukocyte adhesion and recruitment into the vessel wall and surrounding tissue. Cardiovascular disease in patients with vasculitis, although relatively rare, can be life-threatening. Aortitis, hypertension, coronary arteritis, valvular heart disease, pericarditis, myocarditis, conduction abnormalities, accelerated atherosclerosis, and cardiac failure can all occur. This section focuses on the vasculitides most likely to be encountered by cardiovascular disease specialists. GCA affects large and medium-sized arteries. The disease affects those older than 50 years, with incidence increasing with age. GCA occurs most commonly in northern Europe, Scandinavia, and the United States in people of northern European ancestry. GCA typically affects extracranial branches of the aorta and, in addition to the temporal arteries, may involve the subclavian and axillary arteries, the thoracic aorta, and on occasion the femoral and iliac arteries. Clinical features include fever, weight loss, malaise, headache, temporal artery thickening with loss of pulsation, scalp tenderness, and jaw claudication. The most feared complication, anterior ischemic optic neuropathy (AION), may be manifested as amaurosis fugax or sudden permanent visual loss. Up to 25% of patients are initially found to have systemic features without the classic sign of tenderness and temporal artery involvement. 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) has confirmed earlier autopsy findings and shown widespread arteritis with increased FDG uptake throughout the aorta and subclavian and iliac arteries in more than 50% of patients. Histopathologic examination reveals localized fragmentation of the internal elastic lamina closely associated with an inflammatory infiltrate consisting predominantly of IFN-γ–producing CD4+ T lymphocytes, monocytes/macrophages, and occasional characteristic multinucleated giant cells. Recent studies have revealed that activated CD83+ dendritic cells initiate the arterial wall inflammation and colocalize with activated T cells. Local synthesis of growth factors such as platelet-derived growth factor leads to concentric proliferation of smooth muscle cells and occlusion of the arterial lumen (Fig. 84-2). On occasion, release of matrix metalloproteinases and generation of reactive oxygen species result in arterial wall injury and aneurysm formation. Biopsy is the definitive means of diagnosis and should be considered for all patients. However, the need for biopsy should not delay treatment. Temporal artery biopsy is positive in up to 80% of patients. Recent interest has focused on temporal artery ultrasound, which can reveal a characteristic halo sign with concentric homogeneous thickening of the arterial wall and evidence of flow disturbance and stenosis (Fig. 84-2). Although rare, severe cardiovascular complications can occur and include dissecting thoracic aortic aneurysms (Table 84-2). Imaging and autopsy studies suggest that aortitis and aortic wall thickening are frequent in GCA, although their relationship with the development of aortic aneurysm remains unclear. Increased FDG uptake in the thoracic aorta has been associated with an increased risk for aortic dilation.21 Overall, patients with GCA have a 17-fold increased risk for thoracic aortic aneurysms. The risk is higher in those with conventional cardiovascular risk factors, poorly controlled disease, and aortic regurgitation. In the absence of guidelines, we recommend annual thoracic aortic screening for those with FDG-PET–positive thoracic aortic uptake or magnetic resonance angiography (MRA) or computed tomography angiography (CTA) evidence of aortic wall thickening and every 2 to 3 years in the remainder of patients. CTA and MRA are the optimal imaging techniques. Pericarditis, coronary arteritis, limb ischemia, accelerated atherosclerosis, myocardial infarction, and cerebrovascular accidents are all associated with GCA. Yet most outcome studies do not report increased mortality, so the impact of severe cardiovascular disease seems to be small. TABLE 84-2 Cardiovascular Disease in the Systemic Vasculitides IHD = ischemic heart disease; MI = myocardial infarction. TA, a granulomatous panarteritis, affects the aorta and its major branches, typically before the age of 40 years. The disease predominates in women, with a female-to-male ratio of up to 10:1. Because the diagnosis is often delayed, substantial arterial injury accrues. The current diagnostic criteria depend on detection of established stenotic disease and do not yet reflect the increasing sensitivity of noninvasive imaging.22 Initial symptoms typically include nonspecific complaints such as fever, night sweats, arthralgia, malaise, profound tiredness, and lethargy. TA may be accompanied by symptoms of Raynaud phenomenon or upper extremity claudication, and carotidynia occurs in up to 25% of patients. The aorta may be involved throughout its length, and even though any branches can be diseased, the most commonly affected are the subclavian and common carotid arteries. Stenotic/occlusive arterial lesions are found in more than 90% of patients, whereas aneurysms are reported in approximately 25%. The pulmonary arteries are involved in up to 50% of patients, and aortic valve regurgitation and coronary arteritis may occur. The consequences of TA are often severe, with 74% reporting compromised daily activities and 23% unable to work. In our cohort, survival rates at 10 years are higher than 95%; similarly, in the United States, 94% to 96% survival rates are reported, whereas in Korea the survival rate was 87% at 10 years. In Japan, 15-year survival rates have improved to 96.5%. However, the survival rate was reduced to 67% in a subset of patients with serious complications and/or a progressive disease course. Arteritic lesions demonstrate adventitial thickening and focal leukocytic infiltration of the media with intimal hyperplasia. The infiltrate is composed of activated dendritic cells, T and B lymphocytes, macrophages, and multinucleated giant cells (Fig. 84-3). Growth factor–driven myofibroblast proliferation leads to intimal hyperplasia and fibrosis and subsequent arterial stenosis or occlusion. Local matrix metalloproteinase synthesis may predispose to aneurysmal dilation. Diagnosis of TA depends principally on the physician including the disease in the differential diagnosis. The variable nature of the features of TA and the lack of constitutional symptoms in 30% to 50% of patients initially present a challenge to prompt diagnosis. In addition to improved physician awareness, a list of “red flags” that raise the possibility of TA is helpful (Table 84-3).23 One’s index of suspicion must be high in young patients with an unexplained acute-phase response or hypertension. Similarly, common initial signs, including diminished or absent pulsation or arterial bruits, can suggest the diagnosis. Laboratory abnormalities during active disease include raised ESR and CRP (in 75% of patients), often accompanied by normochromic normocytic anemia, thrombocytosis, hypergammaglobulinemia, and hypoalbuminemia. No specific autoantibodies or other serologic abnormalities exist, however. Noninvasive imaging is now the optimal means of diagnosis because tissue biopsy is rarely available. High-resolution ultrasound, cardiac magnetic resonance (CMR), MRA, CTA, and PET have all been studied. Although the potential of these techniques is not in doubt, their specificity and sensitivity in the management of TA remain to be determined. 18F-FDG-PET-CT may reveal evidence of active arteritis and lead to early detection of prestenotic disease. A current consensus review has suggested that this technique is particularly useful for the detection of active arteritis in patients not receiving immunosuppressive therapy.24 Demonstration of arterial wall enhancement, edema, or thickening on MRA and CTA may facilitate the diagnosis of prestenotic disease, and stenoses and aneurysms can be readily identified and monitored. Color duplex ultrasound is of particular use in assessing the common carotid and proximal subclavian arteries in TA. Homogeneous, bright concentric arterial wall thickening is a typical finding in affected common carotid arteries (Fig. 84-3). In addition to the sequelae associated with cerebral, internal organ, and limb ischemia, aneurysms, PAH, or aortic rupture may develop. Cardiac complications include aortic valve insufficiency, accelerated atherosclerosis, cardiac ischemia, myocardial infarction, and heart failure. Neither MRA nor 18F-FDG-PET-CT reliably identifies coronary arteritis. Coronary disease is often asymptomatic, as illustrated by the identification of silent myocardial injury in 27% of a cohort that we studied.25 Patients with TA are also at risk from secondary accelerated atherosclerosis. Thallium stress scintigraphy revealed myocardial perfusion defects in 53%, whereas intra-arterial angiography has shown that up to 30% have coronary artery lesions typically affecting the ostia and proximal segments, with the left main coronary artery being most commonly affected.26 Inflammation of the ascending aorta predisposes to coronary artery involvement, as well as to dilation of the aortic root with subsequent aortic valve regurgitation. In our cohort of 110 patients, 15% have required aortic valve replacement. Left ventricular dysfunction may affect up to 20% and is thought to reflect myocarditis, ischemic heart disease, and hypertension, a common finding often associated with renal artery stenosis in TA. Kawasaki disease (KD) predominantly affects children younger than 5 years with a peak incidence at 6 to 24 months of age. The vasculitis affects medium and small arteries, notably the coronary arteries. All racial groups may be affected, and the highest incidence is recorded in Asia (20 to 100 per 100,000 children <5 years of age). KD is an acute self-limited illness that typically resolves within 1 to 2 months, although mortality still remains 1% to 2%. Characteristic initial features include fever of 5 days’ duration or longer, bilateral conjunctivitis, and mucocutaneous lesions, including red fissured lips and a strawberry tongue. Cervical lymphadenopathy may be prominent, with erythema affecting the palms and soles and a polymorphous exanthema. The cause of KD is unknown, although infection may trigger the disease and lead to an uncontrolled excessive immunologic response in a genetically susceptibly host. The presence of occasional seasonal epidemics and increased incidence in siblings led to the infection hypothesis. A variety of organisms have been implicated, including streptococci, staphylococci, and Propionibacterium acnes. Despite this interest, no definitive evidence supports an infectious cause. Tissue specimens show endothelial injury, perhaps caused by proinflammatory cytokines and activated neutrophils. Infiltration of the arterial wall by neutrophils, T cells, and macrophages is associated with the development of arterial stenosis or, more commonly, aneurysms. Coronary artery aneurysms develop in up to 20% of patients during the first month of the illness, and 50% will regress in the following years. Neutrophilia, thrombocytosis, and a raised acute-phase response occur acutely. Echocardiography can detect coronary involvement from the second week of illness and can be used to monitor progress. Coronary angiography is not performed acutely because of the risk of precipitating myocardial infarction, but it can be used after 6 months to establish the degree of coronary artery injury. The electrocardiogram (ECG) demonstrates abnormalities in up to 50% of patients, including tachycardia, T wave inversion, ST depression, atrioventricular block, and rarely, ventricular arrhythmia. Coronary artery aneurysms develop in up to 25% of untreated patients with KD. Sudden death can occur as a consequence of myocardial infarction following acute coronary thrombosis or rupture of a coronary artery aneurysm. Pericarditis, pericardial effusion, myocarditis, valvular dysfunction, and cardiac failure may all occur, whereas peripheral arterial involvement is less common but may affect the limb, renal, and visceral arteries. Aspirin (80 to 100 mg/kg/day) in four divided doses is recommended, along with intravenous immunoglobulin (IVIG). This treatment combination has reduced the development of coronary artery aneurysm to 5%, with a significant impact on mortality. Twenty percent of cases are resistant to IVIG, however, and these patients can receive corticosteroids, although the results reported are variable. The outcome for most patients with KD is good. Yet in up to 20% of those with coronary artery aneurysms, coronary stenoses eventually develop, and these patients require follow-up by an experienced cardiologist. Although the risk for long-term complications, including myocardial infarction and sudden death, is greater in those with giant aneurysms,27 the risk for thrombosis and myocardial infarction still remains increased in those in whom aneurysms have regressed. Aortitis can be a feature of SLE, Cogan syndrome, Behçet disease, human leukocyte antigen (HLA) B27–positive spondyloarthropathy, KD, and GCA. Aortitis may also be idiopathic in nature. The clinical features are nonspecific and include malaise, lethargy, chest pain, fever, and weight loss, and the diagnosis is often made only at the time of surgery. The ESR and CRP are typically raised, and the extent of the disease can be demonstrated by 18F-FDG-CT-PET scanning and aortic MRA or CTA (Fig. 84-4
Rheumatic Diseases and the Cardiovascular System
Background
Accelerated Atherosclerosis in Rheumatic Diseases
Endothelial Dysfunction and Vascular Injury
Rheumatoid Arthritis
Atherosclerotic Disease in Rheumatoid Arthritis
Treatment
Systemic Lupus Erythematosus
Atherosclerotic Disease in Systemic Lupus Erythematosus
Treatment
Atherosclerosis in Association with Other Rheumatic Diseases
Premature Atherosclerosis
Coronary Arteritis

Vasculitis
Large-Vessel Vasculitis
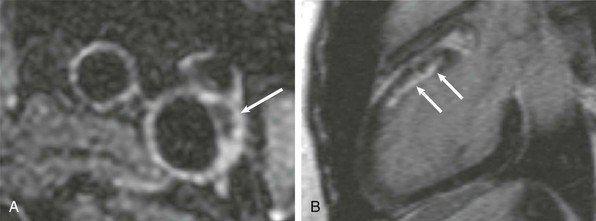
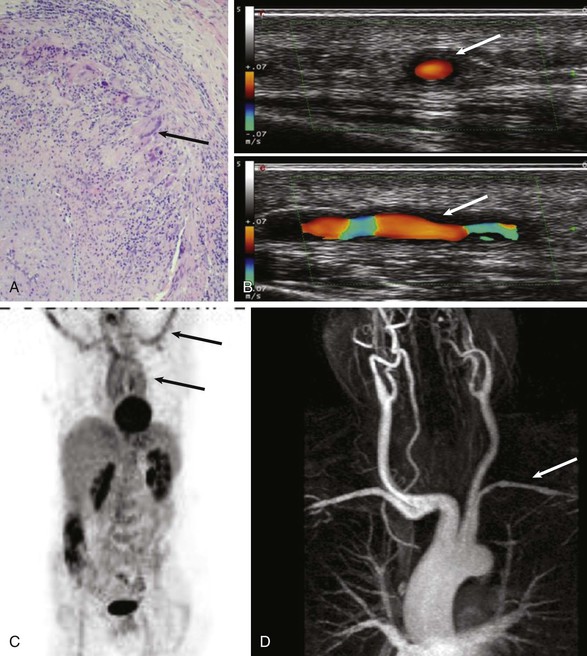
Giant Cell Arteritis
Pathogenesis
Diagnosis
Cardiovascular Complications
VASCULITIDES
CARDIOVASCULAR COMPLICATIONS
Large-Vessel Vasculitis
Giant cell arteritis
Thoracic/abdominal artery aneurysm, limb ischemia, pericarditis, coronary arteritis, IHD, MI
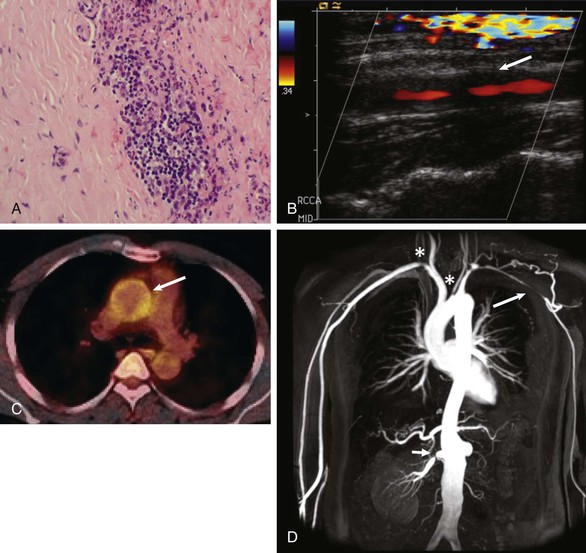
Takayasu arteritis
Aortic regurgitation, limb ischemia, aortic stenosis, aortic aneurysm, stroke, hypertension, coronary arteritis and aneurysm, IHD, MI, myocarditis, cardiac failure
Kawasaki disease
Coronary artery aneurysm, MI, myocarditis, pericarditis, valvular dysfunction, cardiac failure
Medium-Vessel Vasculitis
Churg-Strauss syndrome
Myocarditis, pericarditis, coronary arteritis, cardiomyopathy, cardiac fibrosis, valvular dysfunction, MI
Polyarteritis nodosa
Myocarditis, pericarditis, coronary arteritis, coronary aneurysm, hypertension, cardiac failure
Granulomatous polyangiitis
Myocarditis, pericarditis, coronary arteritis, valvular heart disease, cardiac failure
Microscopic polyangiitis
Pericarditis, coronary microaneurysm, MI
Takayasu Arteritis
Pathogenesis
Diagnosis
Cardiovascular Complications
Kawasaki Disease
Pathogenesis
Diagnosis
Cardiovascular Complications
Treatment
Idiopathic Aortitis
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Rheumatic Diseases and the Cardiovascular System
84