Poor long-term outcomes remain the major clinical challenge in patients hospitalized with acute heart failure (AHF). Although most patients improve symptomatically during the first 24 hours with current medical treatment, their prognosis remains poor with over 15% to 20% mortality and 30% to 40% readmission rate in the first year after discharge. Compared with ambulatory outpatients with heart failure, those hospitalized for AHF have >10-fold increase in the risk of dying, and although this risk decreases exponentially after discharge, it remains twofold to fourfold higher 12 to 18 months later. These outcomes have not changed in the last decade, making AHF one of the most severe clinical conditions leading to hospitalization. Randomized clinical trials have consistently failed to show any improvement in postdischarge outcomes with investigational therapy. This lack of success may because of lack of efficacy, poor drug safety, lack of mechanistic insight, improper matching of drug to a target patient population with many co-morbidities, poor trial execution, or all the previously mentioned. However, another major problem is that, as opposed to other disease entities such as acute coronary syndromes (ACSs), it is difficult for us to identify patients who have clear pathophysiologic abnormalities responsible for the poor prognosis of patients with AHF. Perhaps AHF has such deleterious consequences on patients’ prognosis due to concomitant cardiac, renal, or liver dysfunction that may occur before, during, or after an episode of AHF. Hence, early detection and quantification of organ damage by biomarkers seems a plausible target of treatment, and biomarkers may become valid surrogates of outcomes for patients with AHF.
The current approach to clinical management continues to focus largely on symptom improvement related to congestion. Yet, despite convincing improvement in signs and symptoms, outcomes remain poor. The exact causes for the adverse postdischarge course among these patients are not fully known. A systematic investigation into the reasons for, and downstream complications of, the acute presentation is rarely undertaken. One would not consider removing a small amount of fluid in a patient with pericardial tamponade and alleviate symptoms without considering the cause of the pericardial fluid and ways to prevent it from recurring. This would be similar to treating just the congestion in AHF and not the underlying injury. Further complicating matters is the target of acute therapy, that is the “clot,” may be multifactorial in patients with AHF, and one therapy may not be able to account for the multisystem injury during an AHF presentation. Myocardial injury, as evident by elevated serum cardiac troponin (cTn) levels in many patients with AHF, may mechanistically contribute to such poor outcomes. Recent clinical trial data suggest minimizing injury during an AHF presentation may lead to improved outcomes.
However, not every inpatient with AHF suffers a prolonged hospital stay or organ injury. Differentiating those who will do well and those who are at risk for worsening heart failure (WHF) and subsequent renal or myocardial injury has improved, but further study is needed. Patients may experience this injury before, during, and soon after hospital admission. Patients with troponin elevations during hospitalization have outcomes similar to patients with troponin release at baseline. These elevations may suggest ongoing injury without concomitant clinical symptoms until the postdischarge period and might contribute to the vulnerability of patients during this phase. Such injury during hospitalization may contribute to postdischarge mortality and rehospitalizations. These data provide a compelling argument that the myocardial injury hypothesis should be revisited as future novel pharmacologic agents for AHF are investigated.
Myocyte Adaptations in Chronic and Acute Heart Failure
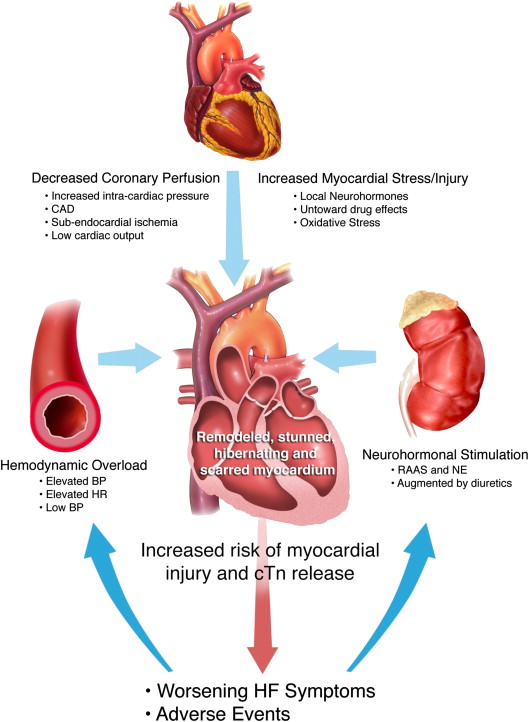
Although the exact pathophysiologic mechanisms leading to myocyte injury in AHF are not fully known, many features of AHF present circumstances for myocyte injury, including (1) excessive neurohormonal stimulation, (2) hemodynamic overload and elevated ventricular end-diastolic pressure, (3) low cardiac output leading to low coronary perfusion and myocardial stress and injury, and (4) tachycardia ( Figure 1 ). This confluence of factors may greatly influence the proportion of patients who suffer myocyte injury and have elevated cTn levels during acute decompensation, particularly those with coronary artery disease. Moreover, AHF therapy may compound this problem, for example with the use of inotropic agents. Compared with patients with heart failure (HF) without elevation, those with cTn elevation are at a significantly increased risk of in-hospital and long-term mortality. A further increase in serum troponin levels during hospitalization is also an independent predictor of long-term outcomes. The mechanism behind cTn release is multifactorial and patients without epicardial coronary artery disease are also at risk for cTn elevations. Importantly, further mechanistic research is needed to identify targets for therapy. It is important to realize that even damaged but viable myocytes may release cTn as a result of increased permeability of the plasma membrane. In patients with HF, cardiac myocytes undergo changes in metabolism to adapt to fluctuations in myocardial oxygen delivery. It has been postulated that the myocytes hibernate to protect themselves from ongoing irreversible myocardial insults, especially during ischemia. Thus patients with chronic HF have both ongoing myocyte turnover, as well as viable, but at-risk, or hibernating, noncontractile myocardium in adjacent areas. The myocyte turnover in these areas of poor oxygen delivery is an additional proposed cause of cTn elevation in AHF.
The percentage of patients with an elevated cTn depends on the severity of HF, the definition of the positive cTn level, and the type of assay used ( Table 1 ). In a large registry analysis, 75% of patients had a detectable cTn above the ninety-ninth percentile, although only 6.2% had values used to define acute myocardial infarction at the time. When a highly sensitive assay is used, nearly all patients with AHF may have a cTn above the ninety-ninth percentile. Up to 70% of patients with ischemic cardiomyopathy may have cTn elevations of >0.3 ng/ml early in their hospital course. Moreover, in patients treated with vasoactive therapy that present initially with undetectable cTn, up to 40% will have detectable levels during hospitalization. Patients who present with an elevated cTn level or subsequently develop an elevation have a significantly higher risk of subsequent rehospitalization or death. This suggests a majority of patients with AHF have ongoing myocardial injury both at the time of hospital presentation and after therapy has been initiated. This pattern may be a significant contributor to the progressive decrease in survival associated with each HF hospitalization.
| Study | N | Definition of cTn elevation | Proportion of AHF patients with cTn elevation at baseline | Outcome: cTn+ vs cTn- groups | |
|---|---|---|---|---|---|
| Perna, 2005 | 184 | cTnT > 0.1 ng/nl | 31.5% | Increased long-term all-cause mortality (p=0.038) | |
| Gheorghiade, 2005 | 51 | 1) cTnT > 0.01 ng/ml 2) cTnI > 0.03 ng/ml | 1) 43% 2) 74% | Increased death or worsening HF at hospital discharge in 1) cTnT (p=0.006) and 2) cTnI (p=0.034) | |
| Sakhuja, 2007 | 209 | cTnT > 0.01 μg/L | 46% | Not prognostic for 1-year mortality unless combined with BNP or NT-proBNP (both p<0.001) | |
| You, 2007 | 2,025 | cTnI > 0.5 μg/L | 34.5% | Increased long-term all-cause mortality (p<0.001) | |
| Peacock, 2008 | 67,294 | 1) cTnT quartiles: 1 st : ≤0.04 2 nd : >0.04-0.10 3 rd : >0.10-0.20 4 th : >0.20 | 2) cTn I quartiles 1 st : ≤0.01 2 nd : >0.01-0.02 3 rd : >0.02-0.06 4 th : >0.06 | 72% of patients had cTnI>0.04 μg/L | Quartiles and associated mortality: 1 st : cTnI (2.0%) cTnT (1.7%) 2 nd : cTnI (2.7%) cTnT (2.8%) 3 rd : cTnI (3.4%) cTnT (3.3%) 4 th : cTnI (5.3%) cTnT (6.3%) |
| O’Connor, 2011 | 288 | 1) Positive: cTnT>0.03 ng/ml 2) Detectable: cTnT>0.01 ng/nl 3) Negative: cTnT≤0.01 ng/ml | 1) 34% 2) 60% 3) 40% | Positive baseline cTnT increased risk of 60-day CV/renal rehospitalization and death (p=0.036) | |
| Metra, 2013 | 1,056 | cTnT > 0.014 μg/L | 93% | Increased 180-day mortality (p<0.0001) | |
In summary, myocardial injury is present during AHF exacerbations in a large proportion of patients with AHF and current therapy may further contribute to it. Troponin, a direct marker of myocardial injury, has been associated with an increased risk of both short- and intermediate-term adverse outcomes. However, whether alteration of a cardiac injury biomarker trajectory with early therapy will alter patient outcomes needs to be studied further.
Unmet Need: Impact of Early Therapy on Myocardial Injury and Subsequent Outcomes
Results from 2 large-scale randomized phase III trials suggest the trajectory of cTn elevation after initial therapy in patients with AHF may have an association with short- and long-term adverse outcomes. The biomarker substudy from the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure trial found 50% of patients had a cTnI above the ninety-ninth percentile upper reference limit (0.034 ng/ml). Baseline cTnI status and absolute increases in cTnI during hospitalization were not associated with an increased risk of 30- or 180-day mortality. However, a 20% increase in cTnI was associated with an increased risk of 30-day mortality. In the Efficacy and Safety of Relaxin for the Treatment of Acute Heart Failure (RELAX-AHF) study high-sensitivity TnT (hs-TnT) levels were above the upper reference limit (0.014 mcg/L) in 93% of patients. Baseline hs-TnT levels and increases at 2, 5, and 14 days were associated with increased 180-day all-cause mortality. Patients who received relaxin had a blunted hs-TnT increase and had a decreased risk of 180-day all-cause mortality compared with placebo. A subsequent secondary analysis suggested patients with a baseline TnT >0.045 μg/L experienced a significant decrease in 180-day cardiovascular death when receiving relaxin compared with placebo. Although overall the patients who received relaxin experienced a significantly lower 180-day death rate than those who received standard care alone, these results suggest patients who experience baseline, and perhaps subsequent cTn elevation, may serve as an ideal target for a therapy that confers benefit during this important early window of AHF management.
Minimizing myocyte injury by increasing coronary artery perfusion, decreasing sympathetic tone and neurohormonal activation during AHF therapy, either alone or in concert, may be necessary to improve in-hospital and postdischarge outcomes for these patients. Many patients with chronic HF have a low ejection fraction, and small differences in myocardial injury and ejection fraction may have significant clinical effects. This is in contrast to patients with ACS, who may experience a bigger insult, but have a significant amount of healthy myocardium to assist with cardiac output.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


