20 Restrictive Cardiomyopathy
Etiology and Pathogenesis
A variety of disease states produce the clinical manifestation of a restrictive cardiomyopathic process (Box 20-1). Myocardial fibrosis, myocardial infiltration by specific proteins, endomyocardial scarring, and cardiac muscle hypertrophy all may present as diastolic dysfunction.
Box 20-1 Classification of Restrictive Cardiomyopathy
With permission, modified from Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–276. (Copyright Massachusetts Medical Society. All rights reserved.)
Noninfiltrative Causes
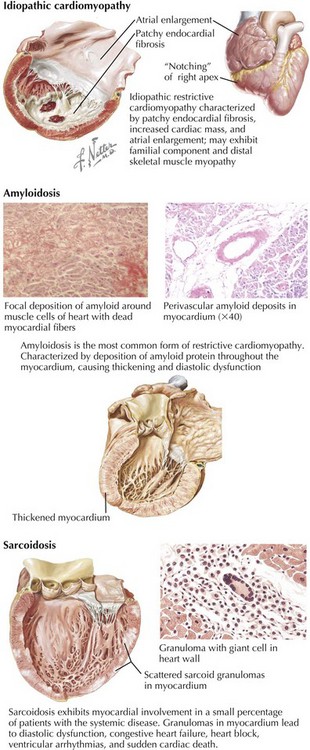
Idiopathic restrictive cardiomyopathy is associated with patchy endomyocardial fibrosis, increased cardiac mass, and enlarged atria (Fig. 20-1). It is more common in older adults but may be seen in children. In adults, 5-year survival is approximately 64%, but mortality may be higher in children. Occasionally, the cardiomyopathy is accompanied by skeletal muscle myopathy, and in some patients with restrictive cardiomyopathy, a clear familial component is present. Idiopathic restrictive cardiomyopathy is also found in families with no skeletal muscle involvement, however, and as an autosomal-dominant disorder in patients with Noonan’s syndrome. Conduction system disease such as atrioventricular (AV) block may also be present and in some cases precedes clinical myocardial dysfunction.
Infiltrative Causes
Clinically, the most common variety of restrictive cardiomyopathy is from amyloidosis, the deposition of unique, twisted, β-pleated sheets of fibrils formed by various proteins (Fig. 20-1, middle). Cardiac amyloidosis can be present in several different circumstances. Primary amyloidosis is caused by deposition of an amyloid protein composed of portions of an immunoglobulin light chain (designated AL for light chain–associated amyloidosis) produced by a monoclonal population of plasma cells. Primary amyloidosis can be the consequence of multiple myeloma but it is also found in patients without multiple myeloma. Secondary amyloidosis, sometimes called reactive systemic disease, is caused by the production of a nonimmunoglobulin protein and termed AA (for amyloid-associated). Familial amyloidosis is an inherited autosomal-dominant trait resulting from a variant prealbumin protein, transthyretin. More than 80 point mutations have been described, and familial amyloidosis may present as a cardiomyopathy, a progressive neuropathy, or a nephropathy. It is four times more common in blacks than in whites. In some cases, the heart is the only affected organ. Senile systemic amyloidosis is produced by an atrial natriuretic-like protein or transthyretin. Its frequency increases with age. Scattered amyloid deposits in the aorta or the atria are almost universally found in individuals older than 80 years, whereas only a small minority of the elderly has evidence of restrictive cardiomyopathy due to amyloidosis.
Sarcoidosis is a granulomatous disease of unknown cause (Fig. 20-1, lower). Of the multiple organ systems commonly involved, including the heart, the most important is usually the lungs, where this involvement manifests as diffuse scarring, pulmonary hypertension, and cor pulmonale. Myocardial involvement causes a restrictive or a dilated cardiomyopathy in less than 5% of systemic sarcoidosis patients. More commonly, focal involvement may result in heart block, congestive failure, ventricular arrhythmias, or sudden cardiac death. The noncaseating granulomas have a propensity for involving the interventricular septum (hence the high incidence of heart block) and the LV free wall. The scattered nature of granulomas contributes to the failure of right ventricular (RV) biopsies to detect the disease in about half the patients. MRI is much more sensitive for detection of cardiac involvement in patients with known sarcoidosis.
Endomyocardial Causes
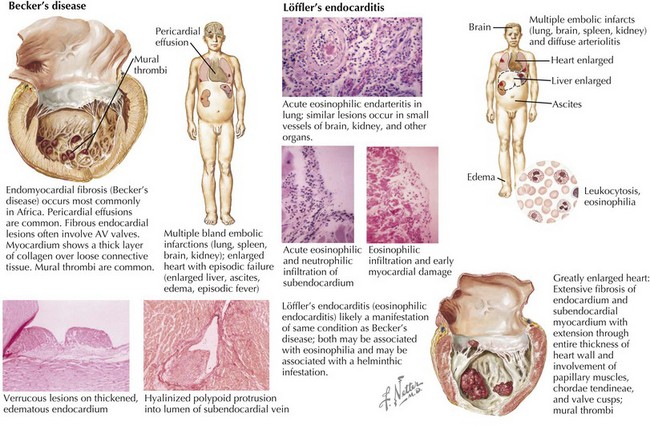
Endomyocardial fibrosis (sometimes called Becker’s disease) occurs most commonly in Africa, especially in Uganda and Nigeria (Fig. 20-2, left). In equatorial Africa, it is responsible for 10% to 20% of deaths from heart disease. Pericardial effusions are common and may be large. Fibrous endocardial lesions are frequently noted in the ventricular inflow tracts and often involve the AV valves, resulting in valvular regurgitation. The involved myocardium demonstrates a thick layer of collagen tissue overlying a layer of loosely arranged connective tissue. Fibrous and granulomatous tissue may extend into the myocardium. Either or both ventricles may be involved, and when the disease process is extensive, papillary muscles and chordae may be matted with a mass of thrombus and tissue, filling the cavity. Clinical manifestations depend on the extent of involvement of the right ventricle, the left ventricle, or both. Eosinophilic endocarditis (Löffler’s endocarditis) is probably an earlier manifestation of this same process (Fig. 20-2, lower). Both diseases are associated with eosinophilia. Epidemiologic evidence suggests that Löffler’s endocarditis is related to worm (helminth) infestation. It is thought that it is during the initial (necrotic) phase of hypereosinophilia that myocardial damage occurs. This is then followed after a year or more by a thrombotic phase and finally a fibrotic, restrictive phase. Clinically, the initial phase is characterized by fever, weight loss, rash, and congestive heart failure (CHF). Localized thickening of the posterolateral LV wall and limited mitral valve movement may be noted. In some instances, the LV apex is virtually obliterated by thrombus. Later, a restrictive pattern with AV regurgitation dominates the hemodynamics, and pericardial effusions, sometimes quite large, are seen.
Other Causes
Hypertrophic cardiomyopathy can present in a similar manner as a restrictive cardiomyopathy. Many mutations in sarcomeric proteins have been identified in genetic studies of hypertrophic cardiomyopathy (see Chapter 19), and there is variability in the degree of diastolic dysfunction depending on both the genotype as well as concomitant diseases (hypertension, diabetes). Generally, it is not difficult to distinguish hypertrophic cardiomyopathy from other causes of restrictive cardiomyopathy.
Differential Diagnosis
Normal Hemodynamics
Intracardiac pressures are a reflection of the contraction and relaxation of individual cardiac structures and the changes imparted to them by the pleural and pericardial pressures (Fig. 20-3). Changes in either pleural or pericardial pressure can be reflected in the intracardiac pressure. With inspiration, the intrapleural pressures drop and the abdominal cavity pressure increases. Blood flow through the right side of the heart increases, whereas blood return to the left side of the heart decreases slightly. The fall in the intrapleural pressures with inspiration also increases the transmural aortic root pressure, effectively increasing the impedance to LV ejection. The reverse occurs during expiration. Normally, inspiration lowers the right atrial and the systolic RV pressures slightly more than it lowers the left heart pressures. In severe lung disease, such as asthma, left heart filling is more profoundly affected, and these changes are exaggerated. The very negative inspiratory intrapleural pressures and very positive expiratory pressures result in marked swings in LV filling. A paradoxical pulse (fall in systemic pressure with inspiration) may thus result from lung disease alone.
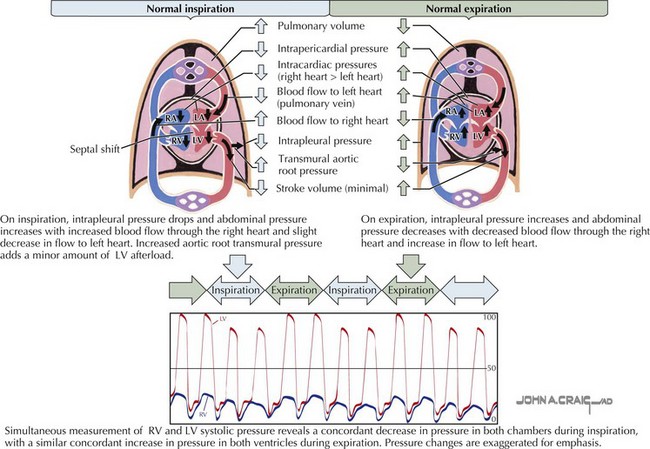
Figure 20-3 Normal cardiac blood flow during inspiration and expiration.
LA, left atrium; LV, left ventricle/ventricular; RA, right atrium; RV, right ventricle/ventricular.
The normal atrial and ventricular waveforms are shown in upper Figure 20-4. With atrial contraction, the atria become smaller and the atrial pressures rise (a wave). With the onset of ventricular contraction, the AV valves bulge toward the atria, and a small c wave is typically detectable on hemodynamic tracings. Although many findings can be seen by careful inspection of the jugular veins on physical examination, the c
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


