32 Restenosis
Re-stenosis is the arterial wall healing response to mechanical injury, which has plagued cardiologists since the introduction of balloon angioplasty by Gruntzig and collaborators.1 This chapter will describe the clinical features, mechanisms, and new facets of BMS and DES re-stenosis in contemporary percutaneous coronary intervention (PCI).
 Definitions
Definitions
Angiographic Re-stenosis
Angiographically detected obstruction of 50% diameter stenosis (DS) or more at the site of a previously treated coronary segment has been historically defined as re-stenosis.2 This apparently arbitrary cut-off point was, in fact, founded on good scientific evidence from physiologic experimental studies, which demonstrated that when the arterial lumen diameter is reduced to 50% or less, the coronary flow reserve becomes impeded.3 For purposes of scientific studies, many definitions of angiographic re-stenosis have been used, although the classic binary definition based on percentage DS is the most widely accepted. Unfortunately, percentage DS does not depict the degree of deterioration in lumen diameter and does not convey a measure of the vessel response to injury.4,5 The use of the term percentage DS itself carries with it the assumption of normal-appearing reference segments, which is known to be an erroneous assumption, given the diffuse aspect of coronary disease and neointimal proliferation.6 Finally, binary re-stenosis assumes that a patient with 51% DS and another one with 49% DS have different intimal hyperplasia responses and outcomes, which, of course, is not the case.
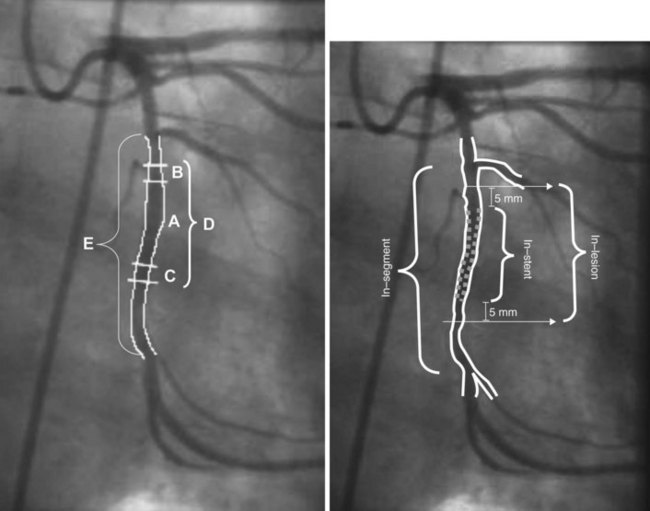
In view of these considerations, clinical re-stenosis studies have been adopting a more comprehensive approach to reporting findings from both perspectives (categorical and continuous) to determine whether the agent under investigation had restraining or inhibitory effect and whether the ultimate clinical or angiographic outcome has been improved by the use of any new therapy. However, the more subtle facets of potent anti-proliferative devices such as DESs challenge the validity of conventional angiographic parameters. Advanced imaging techniques such as intravascular ultrasound (IVUS) and optical coherence tomography (OCT) have greatly improved the ability to visualize re-stenosis and make quantitative assessments of neointimal thickness, neointimal volume, and minimal luminal diameter (MLD).7–12 Late loss (LL) is a continuous angiographic measure of lumen deterioration. LL is conventionally calculated by subtracting the MLD value at follow-up (FU) from postprocedural MLD. These computations are made irrespective of the locations of MLD measurements. LL has traditionally served as a major outcome measure in BMS trials and continues to play a similar role in the era of DESs.13,14 It represents a surrogate marker of neointimal hyperplasia when measurements are performed within stented segments because stents abolish the remodeling component of the re-stenotic process (see below).4,15 However, conventional LL measurements may be methodologically flawed because of changes in the location of MLD between postprocedure and follow-up measurements.9,16 As a result, LL frequently derives from measurements performed in different (unmatched) locations in the target segment. Sites with higher degrees of lumen deterioration and neointimal proliferation may be overlooked, which potentially leads to inaccurate conclusions about the anti-proliferative efficacy of a given device. Angiographic re-stenosis parameters have been reported in two ways: in-lesion analysis and in-stent analysis (Fig. 32-1). In-lesion analysis encompasses the stented segment 5 mm distally and proximally in an attempt to depict edge re-stenosis. It is important to note that in-lesion LL will not necessarily be higher than in-stent LL. These somewhat confusing data occur because of the MLD relocation phenomenon. Furthermore, in-lesion LL is affected by vascular remodeling or vessel spasm and cannot be used as a surrogate for intimal hyperplasia or to determine anti-proliferative device efficacy.
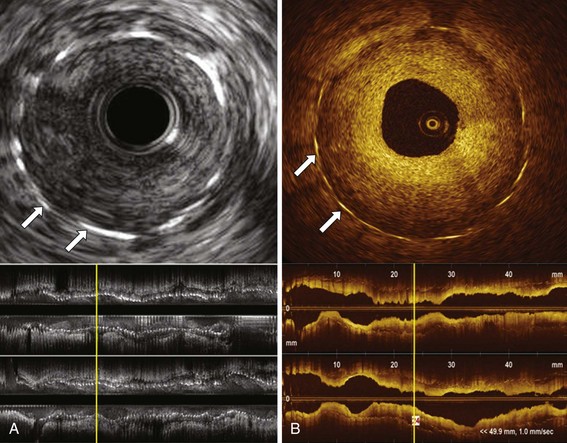
Quantitative coronary angiography (QCA) has been largely used to determine re-stenosis parameters in the clinical context, since visual assessment may lead to overestimation of the degree of narrowing in “severe” lesions and underestimation of the severity in “mild or moderate” lesions.17,18 Clinical and research assessment of re-stenosis has also been markedly improved by IVUS and OCT, which enable high-fidelity measurement of re-stenotic areas, neointimal volume, and MLD, in addition to enabling three-dimensional rendering of the re-stenotic segments. OCT may be a useful technology for the evaluation of stent healing. By using light rather than ultrasound, OCT produces high-resolution in vivo images of coronary arteries and deployed stents with 10 to 15 µm resolution compared with the 150 to 200 µm resolution of IVUS. Although limited in its ability to image deep into the blood vessel wall, OCT increases the diagnostic accuracy of luminal atherosclerosis imaging, thus enabling visualization of small structures such as the thin fibrous caps associated with plaque rupture; it also increases the diagnostic accuracy of stent and re-stenosis imaging, enabling high-resolution assessment of lumen features and neointima (Fig. 32-2).12 In initial clinical trials, OCT outperformed IVUS in the detection of small degrees of neointimal hyperplasia and was more sensitive in detecting stent malapposition.19,20 In addition, OCT-based single-stent, strut-level analysis provides clear assessment of stent–strut coverage and apposition, which are important clinical parameters that have been linked to DES-induced delayed arterial healing and risk of stent thrombosis.21–23
Clinical Re-stenosis
Although angiography has been widely used as the guiding tool in the management of coronary artery disease (CAD), the clinician should also consider functional, invasive or noninvasive fractional flow reserve, assessment of the re-stenotic lesion before referring the patient to additional coronary revascularization. Gruntzig et al observed that most clinical ischemic events related to vessel renarrowing occurred between 3 and 9 months after balloon angioplasty. This seminal observation illustrates the delay between the biologic process and the symptomatic presentation of re-stenosis, which results in a 70% increase in the incidence of repeat revascularization between 6 and 12 months after the procedure.24,25 Potent anti-proliferative strategies may delay the biologic response to injury and extend the time frame to develop clinical signs of re-stenosis. Indeed, intimal proliferation following brachytherapy seems to have a different time course.26 Likewise, re-stenosis after DES may be delayed, although a late catch-up re-stenotic phenomenon has not been observed in clinical studies.27,28 Re-stenosis may cause no symptoms in up to 50% of patients, although silent ischemia may be present.29 Exercise electrocardiographic testing has limited value to detect “silent” re-stenotic lesions, and the detection of re-stenosis by computed tomographic angiography (CTA) can be unreliable because of stent metal radiographic artifacts.30–32 Other noninvasive tests such as thallium scintigraphy and cardiac and stress echocardiography have been used to improve the sensitivity and specificity of noninvasive assessment of re-stenosis.33 On the other end of the clinical spectrum, re-stenosis may present itself in the form of acute coronary syndrome (ACS) in up to one third of patients, which challenges the notion that re-stenosis is a benign condition.34,35 Re-stenosis is of particular concern following left main PCI because of the potential risk of sudden cardiac death (SCD) associated with early “silent” re-stenosis. Despite this theoretical risk, after reviewing clinical data, the writing group for the 2009 Focused Update: ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention decided that the prior class IIa recommendation for angiographic follow-up after left main PCI should be omitted from the 2009 updated guidelines. In making this recommendation, the writing group focused on the inability of angiography to predict a situation that might be prone to acute, sudden stent thrombosis, as well as the risk associated with angiography in a patient who has undergone placement of a left main stent.36 Target lesion revascularization (TLR), defined as any repeat PCI of the treated coronary segment or bypass surgery of the target vessel, has been proposed as the most specific clinical re-stenosis endpoint among other clinical markers (i.e., death, myocardial infarction [MI], symptoms recurrence, or combined major adverse cardiac events [MACEs]).4 Target vessel revascularization (TVR) expands the definition of TLR to include repeat PCI of the target vessel, irrespective of the location of the stenosis within the treated segment. In routine clinical practice, noninvasive assessment of recurrence of re-stenosis (symptomatic status and ischemia tests) appears an appropriate approach. This latter recommendation is based on a series of previous observations: (1) Routine angiographic follow-up may have increased morbidity and mortality, albeit to a small extent; (2) asymptomatic patients with nonfunctional angiographic re-stenosis experience a benign course; and (3) the so-called occulo-stenotic reflex leads to a higher rate of repeat revascularization with no clear clinical benefit at 12 months after the initial intervention.37,38
 Mechanisms of Re-Stenosis
Mechanisms of Re-Stenosis
The pathophysiology of re-stenosis is characterized by neointimal proliferation and negative vascular remodeling. The latter contributes only to re-stenosis after PCI without stent implantation, as the scaffolding properties of stents abolish the remodeling process. It is, nevertheless, important to note that the proliferative vascular response is enhanced by the persistent stimuli of rigid metallic struts in the vessel wall. Neointimal hyperplasia has been originally proposed as a general wound healing response.39 Platelet aggregation, inflammatory cell infiltration, release of growth factors, medial smooth muscle cell (SMC) modulation and proliferation, proteoglycan deposition, and extracellular matrix remodeling were identified as the major milestones in the temporal sequence of this response. It is now recognized that inflammation plays a key role in promoting SMC proliferation, which is central to the pathobiology of re-stenosis.
Smooth Muscle Cell Proliferation and Re-stenosis
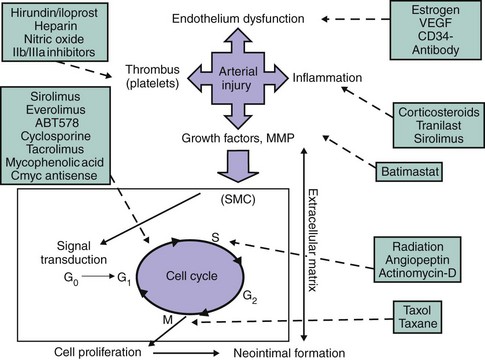
The SMC has long been implicated in the healing process after arterial injury because of its ability to migrate, proliferate, and synthesize extracellular matrix (ECM) on stimulation. After shifting from the contractile to the synthetic phenotype, SMCs may proliferate from 24 hours to 2 to 3 months after vascular injury, returning to the contractile phenotype after this period. Adventitial myofibroblasts (an α-actin staining cell) also proliferate and migrate into the neointima and appear to play an important role in supplying the intimal layer with proliferative cellular elements for new lesion formation.40 Through fracture of the internal elastic membrane, these adventitial cells migrate into the intima, where they may continue to proliferate and synthesize ECM, which will ultimately constitute the bulk of the re-stenotic lesion. ECM is composed of various collagen subtypes and proteoglycans and actually constitutes the major component of the re-stenotic lesion; neointimal hyperplasia has been shown to be predominately a low-cellular tissue.41,42 Assessment of cellular proliferation status in atherectomy specimens of re-stenotic lesions has been enabled by use of antibodies to proliferating cell nuclear antigen (PCNA), cyclin E, and cyclin-dependent kinase (CDK), and there is evidence that cells of monocyte or macrophage lineage (HAM-56–positive) proliferate within human in-stent re-stenotic tissue.2,43,44 The central role of vascular cells in the re-stenotic process provided the basis for anti–re-stenosis pharmacologic strategies targeting cell cycle division early after stent implantation (Fig. 32-3).
Cell Cycle and Re-stenosis
The cell cycle is a common hub of the different phases of the re-stenosis process. The unprecedented clinical successes of recent anti–re-stenosis approaches targeting cellular division pathways illustrate its central role in the formation of neointimal hyperplasia. Currently available DES technologies deliver high concentrations of immunosuppressive or anti-tumor agents locally into the vessel wall. The specific molecular and cellular effects of these agents have been discussed in detail elsewhere.7 SMCs are quiescent and exhibit very low levels of proliferative activity. However, mechanical injury triggers the progress of SMCs through the G1 or GS transition of the cell cycle.45 The different phases of the cell cycle of eukaryotic cells are regulated by a series of protein complexes composed of cyclins (D, E, A, B), CDKs (CDKs, CDK4, CDK2, p34cdc2), and cyclin-dependent inhibitors (CKIs, p27Kip1, p70, p16 INK4). The function of CKIs is regulated by changes in their concentration as well as in their localization in the cell.46 p27Kip1 is downregulated after arterial injury when cell proliferation increases. p21Cip1 is not observed in normal arteries but is upregulated along with p27Kip1 in the later phases of the arterial healing response and is associated with a significant decline in cell proliferation and an increase in procollagen and transforming growth factor beta (TGF-β) synthesis.46 These findings suggest that p27Kip1 and p21Cip1 are endogenous regulators of G1 transit in vascular SMCs and inhibit cell proliferation after arterial injury. p27Kip1 and p21Cip1 bind and alter the activities of cyclin D–, cyclin E–, and cyclin A–dependent kinases (CDK2) in quiescent cells, leading to failure of G1 or GS transition and cell cycle arrest.47,48 Overexpression of p27Kip1 results in cell cycle arrest in the G1 phase. Conversely, inhibition of p27Kip1 increases the number of cells in the GS phase.49 Consistent with its regulatory role in SMC proliferation and the pathobiology of re-stenosis, polymorphisms in p27Kip1 that enhance the production of this cell cycle inhibitor are associated with reduced SMC proliferation and decreased risk of re-stenosis following PCI.50
Inflammation and Re-stenosis
The central role of autocrine and paracrine inflammatory mediators on SMC proliferation is generally well accepted. Leukocyte recruitment and infiltration occur early at sites of vascular injury, where the lining endothelial cells have been denuded and platelets and fibrin have been deposited. The initial tethering and rolling of leukocytes on platelet P-selectin are followed by their firm adhesion and trans-platelet migration, processes that are dependent on leukocyte Mac-1 and platelet glycoprotein (GP) Ibα.51–53 The precise cellular and molecular mechanisms of inflammation following arterial injury are highly dependent on the specific type of injury (i.e., stent versus balloon mechanical versus atherogenesis). Experimental stent deployment in animal arteries causes sustained elevation of monocyte chemoattractant protein-1 (MCP-1) after injury (~14 days) compared with balloon-injured arteries (<24 hours).54 Antibody-mediated blockade of chemokine receptor 2 (CCR-2), a primary leukocyte receptor for MCP-1, markedly diminished neointimal thickening after stent-induced injury, but not balloon-induced injury, in nonhuman primates.55 Experimental observations support a causal relationship between inflammation and experimental re-stenosis. Antibody-mediated blockade or selective absence of Mac-1 diminished leukocyte accumulation and limited neointimal thickening after experimental angioplasty or stent implantation. Corticosteroids have long been shown to reduce the influx of mononuclear cells, to inhibit monocyte and macrophage function, and to influence SMC proliferation.56–59 However, clinical trials with systemic steroid therapy to prevent re-stenosis have shown disappointing results.60
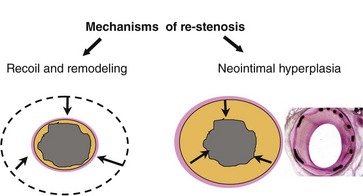
Remodeling and Re-stenosis
The term remodeling has been applied largely to describe either vascular shrinkage or vascular enlargement. The definition proposed by Schwartz et al, in which remodeling is characterized in a continuous spectrum by any change in vascular dimension, may better describe this compensatory phenomenon.61 Studies using intravascular ultrasound (IVUS) provided the first evidence of the key role of negative remodeling (vessel shrinkage) on lumen deterioration after nonstented PCI.62,63 Adventitial myofibroblasts, which are capable of collagen synthesis and tissue contraction, as seen in wound healing, may play an important role in the negative vessel remodeling observed in re-stenosis after balloon angioplasty.64,65 Nevertheless, remodeling is virtually absent after stenting as observed by volumetric IVUS (Fig. 32-4).66,67 The superior outcomes of BMSs compared with angioplasty result mainly from the scaffolding property of these metallic prostheses, which prevents vessel shrinkage (elastic recoil and negative remodeling) despite inducing an enhanced neointimal hyperplasia response.
Specific Mechanisms of Bare Metal Stent Re-stenosis
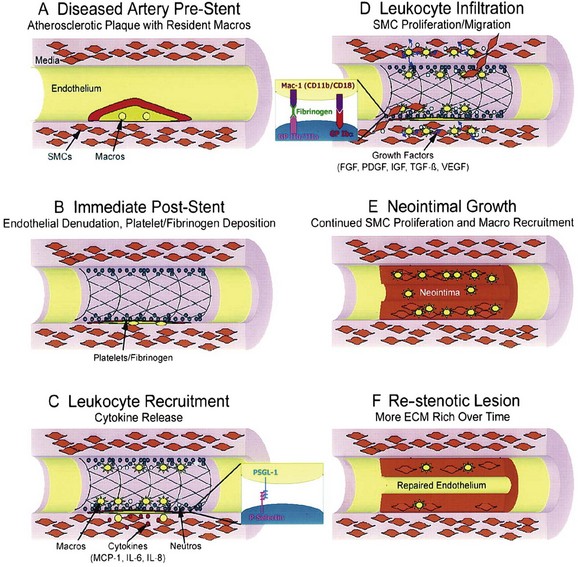
The initial consequences immediately following stent placement are de-endothelialization; crush of the plaque often with dissection into the tunica media and, occasionally, adventitia; and stretch of the entire artery.68 A layer of platelets and fibrin is then deposited at the injured site. Activated platelets on the surface expressing adhesion molecules such as P-selectin attach to circulating leukocytes via platelet receptors such as P-selectin glycoprotein ligand (PSGL-1) and begin a process of leukocyte rolling along the injured surface. Leukocytes then bind tightly to platelets through the interaction of leukocyte Mac-1 with platelet GP Ibα and through leukocyte integrin cross-linking with fibrinogen, which is bound to the platelet GP IIb/IIIa receptor. Migration of leukocytes across the platelet–fibrin layer and diapedesis into the tissue are driven by the chemical gradients of chemokines released from SMCs and resident macrophages. This is followed by a granulation or cellular proliferation phase. Growth factors are subsequently released from platelets, leukocytes, and SMCs and stimulate the migration of SMCs from the media and adventitia into the intimal layer. The resultant neointima consists of SMCs, ECM, and macrophages recruited over a period of several weeks. Cellular division takes place in this phase, which appears to be essential for the subsequent development of re-stenosis. Over a longer period, the artery enters a phase of remodeling involving ECM protein degradation and resynthesis (Fig. 32-5). Accompanying this phase is a shift to less cellular elements and greater production of ECM. In stented arteries, re-endothelialization of the surface of the stented vessel occurs in parallel with the proliferative processes described above.
Specific Mechanisms of Drug-Eluting Stent Re-stenosis
Despite almost a decade of clinical experience with DES technologies in human PCI, current understanding of the mechanisms of DES re-stenosis remains somewhat limited. Compared with BMS re-stenosis, the biology of re-stenosis is probably altered by the potent anti-proliferative agents released from DES devices. The cellular composition of human DES re-stenotic tissue may vary from a T lymphocyte–prominent cellular infiltrate to a predominantly SMC- and ECM-rich tissue similar to BMS re-stenosis. Several lines of evidence suggest that vascular responses, in general, are altered by DESs compared with BMSs. An echolucent intimal hyperplasia, named black hole, has been identified by IVUS in patients treated with DESs (Fig. 32-6).69,70
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


