Respiratory Failure: An Overview
Respiratory failure is a condition in which the respiratory system fails in one or both of its gas-exchanging functions; that is, oxygenation of, and carbon dioxide elimination from, mixed venous (pulmonary arterial) blood. Hence, respiratory failure is a syndrome rather than a disease. Many diseases result in respiratory failure, as discussed elsewhere in this volume.
Respiratory failure may be acute or chronic. The clinical presentations of patients with acute and chronic respiratory failure usually are quite different. While acute respiratory failure is characterized by life-threatening derangements in arterial blood gases and acid–base status, the manifestations of chronic respiratory failure are more indolent and may be clinically inapparent.
Although the causes of respiratory failure are diverse, common underlying pathophysiological mechanisms and management strategies merit a general discussion. This chapter begins with a focus on the definition of respiratory failure and underscores distinctions between acute and chronic varieties. Hypoxemic and hypercapnic respiratory failure are described, and the pathophysiological underpinnings of each type are reviewed. The concepts of ventilatory supply and demand are considered before an overview of the many categories of disease that result in respiratory failure. Finally, an approach to clinical evaluation and management is outlined, followed by a summary of complications, comments on prognosis, and consideration of sites of care for survivors of respiratory failure who remain “chronically critically ill.”
CLASSIFICATION OF RESPIRATORY FAILURE
As noted previously, respiratory failure is characterized by inadequate blood oxygenation or carbon dioxide removal. “Adequacy” is defined by tissue requirements for oxygen uptake and carbon dioxide elimination. In the absence of bedside techniques for direct measurement of these metabolic parameters, clinicians must rely on arterial blood gas values.
Respiratory failure may be classified as hypercapnic or hypoxemic (Fig. 139-1). Hypercapnic respiratory failure is defined as an arterial PCO2 (PaCO2) greater than 45 mm Hg. Hypoxemic respiratory failure is defined as an arterial PO2 (PaO2) less than 55 mm Hg when the fraction of oxygen in inspired air (FIO2) is 0.60 or greater. In many cases, hypercapnic and hypoxemic respiratory failure coexist. Disorders that initially cause hypoxemia may be complicated by respiratory pump failure (see below) and hypercapnia. Conversely, diseases that produce respiratory pump failure are frequently complicated by hypoxemia due to secondary pulmonary parenchymal processes (e.g., pneumonia or atelectasis) or vascular disorders (e.g., pulmonary embolism).
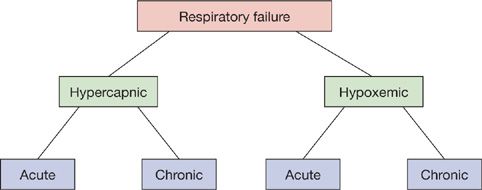
Figure 139-1 Classification of respiratory failure. Although depicted as distinct entities, hypercapnic and hypoxemic respiratory failure frequently coexist. Either may be acute or chronic.
Distinctions between acute and chronic respiratory failure are summarized in Table 139-1. In general, acute hypercapnic respiratory failure is defined as a PaCO2 greater than 45 mm Hg with accompanying acidemia (pH less than 7.30).1 The physiological effect of a sudden increment in PaCO2 depends on the prevailing level of serum bicarbonate anion. In patients with chronic hypercapnic respiratory failure – for example, due to chronic obstructive pulmonary disease (COPD) – a long-standing increase in PaCO2 results in renal “compensation” and an increased serum bicarbonate concentration. A superimposed acute increase in PaCO2 has a less dramatic effect than does a comparable increase in a patient with a normal bicarbonate level.
TABLE 139-1 Distinctions Between Acute and Chronic Respiratory Failure
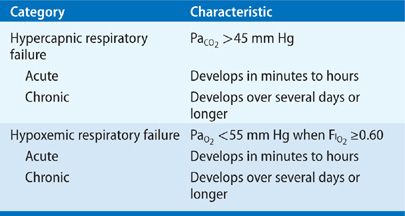
Distinction between acute and chronic hypoxemic respiratory failure may not be readily made on the basis of arterial blood gas values.1 The presence of markers of chronic hypoxemia (e.g., polycythemia or cor pulmonale) provides clues to a long-standing disorder, whereas abrupt changes in mental status suggest an acute event.
An important fact to bear in mind is that even though the definition of hypoxemic respiratory failure rests on measurement of PaO2, the major threat of arterial hypoxemia is inadequate tissue oxygenation, reflected in tissue oxygen delivery. Tissue oxygen delivery is determined by the product of cardiac output and blood oxygen content (see Chapter 15, Blood-Gas Transport); the latter, in turn, depends on hemoglobin concentration and oxygen saturation. Therefore, factors that lower cardiac output or hemoglobin concentration, or inhibit dissociation of oxygen from hemoglobin at the tissue level, may promote tissue hypoxia without technically producing respiratory failure.
PATHOPHYSIOLOGY
Respiratory failure can arise from an abnormality in any of the “effector” components of the respiratory system—central nervous system, peripheral nervous system, respiratory muscles and chest wall, airways, or alveoli (Fig. 139-2). A defect in any of the first four components, which constitute the “respiratory pump,” may cause coexistent hypercapnia and hypoxemia; at least initially, disorders of the alveoli are more apt to result in hypoxemia.2,3
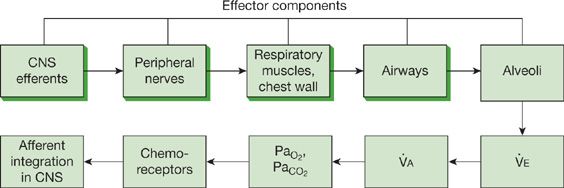
Figure 139-2 Functional components of the respiratory system and its controller. Abnormalities in any of the effector components can result in respiratory failure. The central and peripheral nervous systems, respiratory muscles and chest wall, and airways constitute the “respiratory pump” (shaded boxes). Hypercapnia is the hallmark of respiratory pump failure, while hypoxemia constitutes the primary disturbance in alveolar disorders producing respiratory failure. (Reproduced with permission from Lanken PN. Pathophysiology of respiratory failure. In: Grippi MA, ed. Pulmonary Pathophysiology. Philadelphia. PA: JB Lippincott; 1995.)
 HYPOXEMIC RESPIRATORY FAILURE
HYPOXEMIC RESPIRATORY FAILURE
As described in Chapters 14–16, four pathophysiological mechanisms account for the hypoxemia seen in a wide variety of diseases: alveolar hypoventilation, ventilation–perfusion mismatch, shunt, and diffusion limitation.2 Alveolar hypoventilation occurs in neuromuscular disorders that affect the respiratory system. In the absence of underlying pulmonary disease, the hypoxemia accompanying alveolar hypoventilation is characterized by a normal alveolar–arterial oxygen gradient,4 as defined by Equation 1:
![]()
where
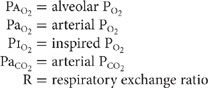
In contradistinction, disorders in which any of the other three mechanisms are operative are characterized by widening of the alveolar–arterial oxygen gradient, which is normally less than 20 mm Hg. With ventilation–perfusion mismatching, areas of low ventilation relative to perfusion contribute to the hypoxemia. Similarly, with shunt, either intrapulmonary or intracardiac, deoxygenated mixed venous blood bypasses ventilated alveoli, resulting in “venous admixture.” Finally, diseases that increase the diffusion pathway for oxygen from the alveolar space to pulmonary capillary impair oxygen transport across the alveolar-capillary membrane.
Although changes in minute and alveolar ventilation can change PaCO2 considerably, this is not so for PaO2. Increases in minute ventilation and, secondarily, in alveolar ventilation modestly increase PaO2. Indeed, at a PaO2 above 55 to 60 mm Hg, the effect of increasing ventilation on oxygen content is minimal, since the oxyhemoglobin dissociation curve is flat in this range.
 HYPERCAPNIC RESPIRATORY FAILURE
HYPERCAPNIC RESPIRATORY FAILURE
At a constant rate of CO2 production (![]() CO2), PaCO2 is determined by the level of alveolar ventilation.5 The relationship between alveolar ventilation, rate of CO2 production, and PaCO2 is described by Equation 2:
CO2), PaCO2 is determined by the level of alveolar ventilation.5 The relationship between alveolar ventilation, rate of CO2 production, and PaCO2 is described by Equation 2:
![]()
where

When ![]() CO2 is constant, PaCO2 is determined by
CO2 is constant, PaCO2 is determined by ![]() A, which, in turn, is dictated by two factors: minute ventilation (
A, which, in turn, is dictated by two factors: minute ventilation (![]() E) and the relationship between
E) and the relationship between ![]() E and
E and ![]() A. The latter is determined by the proportion of
A. The latter is determined by the proportion of ![]() E that constitutes dead space ventilation—that is, the dead space to tidal volume ratio (VD/VT):
E that constitutes dead space ventilation—that is, the dead space to tidal volume ratio (VD/VT):
![]()
where

Inspection of Equation 3 indicates that disorders reducing ![]() E or increasing the proportion of dead space ventilation may result in hypercapnia.
E or increasing the proportion of dead space ventilation may result in hypercapnia.
 VENTILATORY SUPPLY VERSUS DEMAND
VENTILATORY SUPPLY VERSUS DEMAND
A useful theoretical construct for understanding the pathophysiological basis for hypercapnic respiratory failure is the relationship between ventilatory supply and ventilatory demand (Fig. 139-3).1
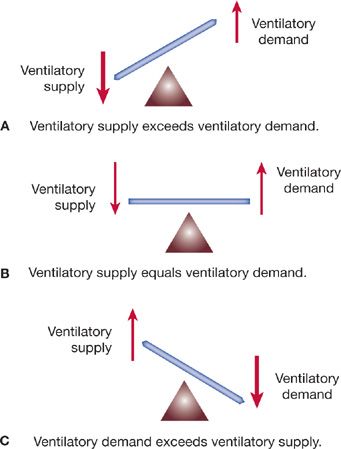
Figure 139-3 Relationship between ventilatory supply (maximal sustainable ventilation) and ventilatory demand (overall level of ventilation specified by the CNS controller). Relative size of the arrows indicates levels of supply and demand in each of the three circumstances illustrated. A. Normal. Ventilatory supply greatly exceeds ventilatory demand. Physiological “reserve” is maintained. B. Ventilatory supply is decreased and ventilatory demand increased (e.g., acute asthma attack). “Borderline” respiratory failure exists. C. Ventilatory demand exceeds ventilatory supply (e.g., sepsis in a patient with COPD). Respiratory muscle fatigue develops, and hypercapnic respiratory failure ensues. See text for details. (Reproduced with permission from Lanken PN. Pathophysiology of respiratory failure. In: Grippi MA, ed. Pulmonary Pathophysiology. Philadelphia, PA: JB Lippincott; 1995.)
Ventilatory supply is the maximal spontaneous ventilation that can be maintained without development of respiratory muscle fatigue; ventilatory supply is also known as maximal sustainable ventilation (MSV).
Ventilatory demand is the spontaneous minute ventilation, which, when maintained constant, results in a stable PaCO2 (assuming a fixed rate of CO2 production).
Normally, ventilatory supply greatly exceeds ventilatory demand. Hence, major changes in minute ventilatory requirements (e.g., during exercise) may occur without hypercapnia. In lung disease, significant abnormalities may be present before ventilatory demand encroaches on MSV. Consequently, hypercapnia is a late finding. When ventilatory demand exceeds MSV, PaCO2 increases.
As a general rule, MSV is approximated as one-half the maximal voluntary ventilation, or MVV (see Chapter 33, Pulmonary Function Testing). A 70-kg adult has an MVV of about 160 L/min, an MSV of 80 L/min, and, under basal conditions, a ![]() E of approximately 6 to 7 liters per minute (90 mL/kg/min). Normally, therefore, there is a 10- to 15-fold difference between resting
E of approximately 6 to 7 liters per minute (90 mL/kg/min). Normally, therefore, there is a 10- to 15-fold difference between resting ![]() E and MSV. In disease states, the
E and MSV. In disease states, the ![]() E requirement may approach a markedly reduced MSV. Further reductions in MSV result in ventilatory demand exceeding supply, and hypercapnia occurs.
E requirement may approach a markedly reduced MSV. Further reductions in MSV result in ventilatory demand exceeding supply, and hypercapnia occurs.
FACTORS THAT REDUCE VENTILATORY SUPPLY OR INCREASE VENTILATORY DEMAND
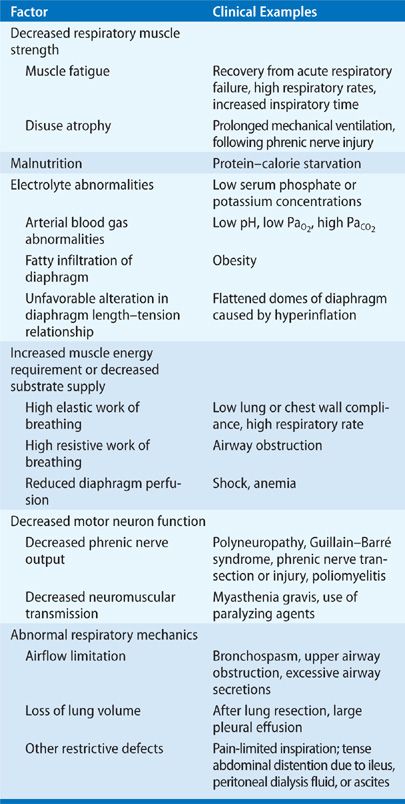
Disruption of any component of the efferent arm of the respiratory control system may diminish ventilatory supply (Table 139-2). While a variety of diseases produce specific abnormalities along the efferent pathway (e.g., phrenic nerve and respiratory muscle disorders), some result in respiratory muscle fatigue—the biochemical, cellular, and molecular mechanisms of which remain poorly understood.
TABLE 139-2 Factors That Diminish Ventilatory Supply
As described previously, ventilatory demand can be assessed according to Equation 3:
![]()
Any factor that affects terms on the right-hand side of the equation may result in ventilatory demand exceeding supply. Selected clinical examples are given in Table 139-3.
TABLE 139-3 Factors That Increase Ventilatory Demand

CATEGORIES OF RESPIRATORY FAILURE
Although many different diseases cause respiratory failure, they may be grouped conveniently according to primary abnormalities in the individual effector components of the respiratory system.1,2
 ABNORMALITIES OF THE CENTRAL NERVOUS SYSTEM
ABNORMALITIES OF THE CENTRAL NERVOUS SYSTEM
A variety of pharmacologic, structural, and metabolic disorders of the CNS are characterized by suppression of the neural drive to breathe.3 The resultant hypoventilation and hypercapnia may be acute or chronic.
An overdose of a narcotic or other drug with sedative properties is a common cause of respiratory failure. While the most striking clinical picture occurs with an acute overdose, long-standing use of some agents (e.g., methadone) may result in chronic hypercapnia.
“Structural” CNS abnormalities producing hypercapnic respiratory failure include meningoencephalitis, localized tumors or vascular abnormalities of the medulla, and strokes affecting medullary control centers. Usually, respiratory failure is observed in the context of other neurologic findings.
A variety of metabolic derangements may produce hypercapnia through depression of respiratory control centers. Examples include severe myxedema, hepatic failure, and advanced uremia. In addition, elevation of PCO2 in the CNS results in neural depression, further enhancing CO2 retention. A common clinical setting in which elevation of PaCO2 is observed is chronic metabolic alkalosis (e.g., due to diuretic use), as detailed in Chapter 17 (Acid–Base Balance).
Finally, obesity hypoventilation syndrome is characterized by hypercapnia due to hypoventilation on a central basis.6
 ABNORMALITIES OF THE PERIPHERAL NERVOUS SYSTEM OR CHEST WALL
ABNORMALITIES OF THE PERIPHERAL NERVOUS SYSTEM OR CHEST WALL
A wide variety of disorders of the peripheral nerves, neuromuscular junction, and chest wall may be associated with hypercapnic and hypoxemic respiratory failure.3,7–16 While the hallmark is an inability to maintain a level of ![]() E appropriate for the rate of CO2 production, many of these disorders are complicated by impaired expiratory muscle strength, atelectasis, and aspiration. Through mechanisms outlined previously, hypoxemia develops in conjunction with the hypercapnia.
E appropriate for the rate of CO2 production, many of these disorders are complicated by impaired expiratory muscle strength, atelectasis, and aspiration. Through mechanisms outlined previously, hypoxemia develops in conjunction with the hypercapnia.
Among the most common neuromuscular causes of hypercapnic respiratory failure are Guillain–Barré syndrome,17 myasthenia gravis,18 polymyositis,19 the muscular dystrophies, amyotrophic lateral sclerosis (ALS),20 and a large number of metabolic muscle disorders. In addition, acute poliomyelitis and traumatic spinal cord injury are associated with hypercapnia. Development of respiratory muscle fatigue and atrophy10,21–23 during prolonged weaning from mechanical ventilation may cause recurrent hypercapnia in the critical care setting.
Pharmacologic causes of hypercapnia in the intensive care unit are frequently encountered. Early reports on use of depolarizing and nondepolarizing paralyzing agents,24 particularly in conjunction with systemic corticosteroids (e.g., in management of status asthmaticus) have been well documented; cholinergic crisis during therapy of myasthenia gravis, and administration of aminoglycosides to patients with myasthenia gravis are additional examples.
Primary disorders of the chest wall constitute another important category of neuromuscular respiratory failure. The prototype is severe kyphoscoliosis. Additional examples include flail chest (Chapter 106, Thoracic Trauma), extensive thoracoplasty, morbid obesity, and massive abdominal distention due to ascites25 or distended loops of bowel.
In each of these disorders, a common pathophysiological sequence develops. Because of inadequate activation of inspiratory muscles or limited thoracic excursion, tidal volume falls. While an increase in respiratory rate compensates initially for the fall in ![]() E (and in
E (and in ![]() A),
A), ![]() E eventually declines. In addition, the sigh mechanism is impaired, which, in conjunction with the low tidal volume, results in atelectasis and reduced lung compliance. Reduced lung compliance produces a further fall in tidal volume and an increase in the elastic work of breathing (Chapter 10, Pulmonary Mechanics). Hence, ventilatory supply becomes limited, while ventilatory demand increases due to a rise in VD/VT (as a result of atelectasis and other factors noted below). An imbalance between ventilatory supply and demand arises, and hypercapnia ensues. Furthermore, an impaired gag reflex in the setting of bulbar weakness, coupled with impaired cough due to respiratory muscle involvement, may result in aspiration pneumonia and secondary hypoxemia.
E eventually declines. In addition, the sigh mechanism is impaired, which, in conjunction with the low tidal volume, results in atelectasis and reduced lung compliance. Reduced lung compliance produces a further fall in tidal volume and an increase in the elastic work of breathing (Chapter 10, Pulmonary Mechanics). Hence, ventilatory supply becomes limited, while ventilatory demand increases due to a rise in VD/VT (as a result of atelectasis and other factors noted below). An imbalance between ventilatory supply and demand arises, and hypercapnia ensues. Furthermore, an impaired gag reflex in the setting of bulbar weakness, coupled with impaired cough due to respiratory muscle involvement, may result in aspiration pneumonia and secondary hypoxemia.
In addition to the pathophysiology described, structural abnormalities of the thoracic cage (e.g., severe kyphoscoliosis, ankylosing spondylitis, thoracoplasty, and fibrothorax)7,26,27 are characterized by an increase in the elastic component of the work of breathing. This results in a higher ![]() O2 and a higher proportion of total O2 consumption by the respiratory muscles (normally, less than 5% of
O2 and a higher proportion of total O2 consumption by the respiratory muscles (normally, less than 5% of ![]() O2).
O2).
 ABNORMALITIES OF THE AIRWAYS
ABNORMALITIES OF THE AIRWAYS
Obstructive diseases of the airways – either upper or lower – are common causes of acute and chronic hypercapnia. Examples in the upper airways include acute epiglottitis, aspirated foreign body, tracheal tumor, and narrowing of the trachea or glottis by fibrotic tissue. Disorders of the lower airways include COPD,28 asthma, and advanced cystic fibrosis. Increasingly, the development of bronchiolitis obliterans, a manifestation of allograft rejection in patients who have undergone lung transplantation, has been recognized as an important cause of respiratory failure.29
The underlying mechanisms in airway abnormalities as causes of respiratory failure are multifaceted and variable. However, several common pathophysiological pathways are operative.
Airway narrowing results in a greater transthoracic pressure gradient requirement for inspiratory airflow. The resistive component of the work of breathing is increased, and the increase is associated with an elevation in ![]() O2. In addition, tidal volume falls and dead space ventilation increases. Respiratory muscle fatigue may develop; the consequences of a shallow breathing pattern ensue.
O2. In addition, tidal volume falls and dead space ventilation increases. Respiratory muscle fatigue may develop; the consequences of a shallow breathing pattern ensue.
Finally, in some disorders (e.g., acute asthma or an acute exacerbation of COPD), air trapping and lung hyperinflation occur, resulting in diaphragm flattening and worsening diaphragm mechanics (see Chapter 10, Pulmonary Mechanics). The overall effect is a growing imbalance between ventilatory supply and demand.1
 ABNORMALITIES OF THE ALVEOLI
ABNORMALITIES OF THE ALVEOLI
Although diseases characterized by diffuse alveolar filling frequently result in hypoxemic respiratory failure, hypercapnia may complicate the picture. Common clinical examples in this category include cardiogenic and noncardiogenic pulmonary edema,30 diffuse pneumonia, extensive pulmonary hemorrhage, aspiration of stomach contents, and near drowning.
Diffuse alveolar filling creates a large right-to-left shunt as pulmonary blood flows through nonventilated or poorly ventilated regions of the lung. In addition, coexisting interstitial edema may impair diffusion across the alveolar-capillary membrane, further impairing oxygenation of mixed venous blood.
In extensive, acute pulmonary disease characterized by alveolar filling, ventilatory demand is high because of hypoxemia and increases in VD/VT, the elastic work of breathing (due to reduced lung compliance), the resistive work of breathing (due to airway narrowing and increased airway reactivity), and the neural drive to breathe (mediated by pulmonary parenchymal vagal fibers). In conjunction with heightened ventilatory demand, ventilatory supply is reduced because of alveolar flooding, reduced lung elasticity, respiratory muscle fatigue, and, possibly, reduced blood supply to the diaphragm secondary to shock. Once again, the imbalance between ventilatory supply and demand results in hypercapnia.
APPROACH TO THE PATIENT
The diagnosis of acute or chronic respiratory failure begins with clinical suspicion of its presence. Confirmation of the diagnosis is based on arterial blood gas analysis. Evaluation for an underlying cause must be initiated early, frequently in the presence of concurrent treatment for acute respiratory failure. While the diagnosis of chronic respiratory failure is usually easily established with clinical findings of chronic hypoxemia (with or without findings of hypercapnia), the diagnosis of acute respiratory failure requires more careful analysis.
Signs and symptoms in acute respiratory failure reflect the underlying disease process and associated hypoxemia or acidemia due to hypercapnia. Localized pulmonary findings reflecting the acute causes of hypoxemia (e.g., pneumonia, pulmonary edema, asthma, or COPD) may be readily apparent. Alternatively, the predominant findings may be systemic (e.g., hypotension due to sepsis). The principal manifestations may even be remote from the thorax – for example, abdominal pain in acute pancreatitis or leg pain due to a long-bone fracture – each associated with acute respiratory distress syndrome (Chapter 140, Acute Respiratory Distress Syndrome: Pathogenesis and Chapter 141, Acute Respiratory Distress Syndrome: Clinical Features, Management, and Outcomes). Frequently, neurologic or cardiovascular symptoms and signs predominate. Neurologic manifestations include restlessness, anxiety, confusion, seizures, or coma. Asterixis may be seen with severe hypercapnia. Common cardiovascular findings include tachycardia and a variety of arrhythmias. Finally, there may be few or no findings other than a complaint of dyspnea, as in some patients with hypoxemia due to pulmonary embolism.
Once respiratory failure is suspected on clinical grounds, arterial blood gas analysis is performed to confirm the diagnosis, to assist in the distinction between acute and chronic forms, to assess the magnitude and metabolic impact, and to help guide management (Table 139-4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


