Regulation of Cardiac Muscle Performance
Regulatory Mechanisms in Cardiac and Skeletal Muscle
Muscle performance can be regulated by the four mechanisms listed in Table 10-1. To meet different physiological and pathological challenges, these are used differently in cardiac and skeletal muscle. In most skeletal muscles, where periods of activity alternate with periods of rest, myocyte load is determined by the mass that must be moved, gravity, and the angles at skeletal joints. In the heart, which contracts without pause, myocyte load is influenced by chamber size and shape as well as by circulatory hemodynamics. This is because geometrical laws determine the relationship between myocyte shortening in the walls of the heart and the volume of blood ejected in each beat, and because wall stress in the heart is determined by chamber volume, as well as by wall thickness and chamber pressure (the law of Laplace, described in Chapter 11). Regulation of cardiac muscle performance also differs from that of most skeletal muscles because the body must meet sustained physiological stresses, such as the need to increase cardiac output during endurance exercise, by activating the functional responses described in Chapter 8. Adaptation of cardiac performance to the more prolonged pathological stresses caused by chronic hemodynamic overload and heart disease is accompanied by activation of the proliferative responses described in Chapter 9.
Summation of Contractions
The tension developed by a skeletal muscle can be increased when rapid stimulation by motor neurons causes summation or tetanic contractions (see Chapter 6); however, these mechanisms are of little physiological importance because most skeletal muscle contractions are brief tetani. Exceptions are the contractions of extraocular muscles, which are often twitches, but these brief contractile responses, which resemble the knee jerk reflex, would not be useful for most movements.
Summation plays no role in the heart because cardiac action potentials normally last almost to the end of the active state. For this reason, an activated cardiac myocyte cannot respond to additional electrical stimuli until relaxation is virtually complete, which makes it impossible for summation and tetanic contraction to be used for physiological regulation.
Variations in the Number of Active Motor Units
Skeletal muscles are composed of groups of myocytes, called motor units, each of which is innervated by a single motor neuron that operates independently of other motor units. This allows performance to be regulated by the central nervous system, which can vary the number
of active motor units. For example, when a muscle is called upon to lift a light load, only a small fraction of its motor units are activated, whereas a greater proportion of the motor units are activated via their motor neurons when the same muscle lifts a heavy load. This mechanism is the major determinant of skeletal muscle performance.
of active motor units. For example, when a muscle is called upon to lift a light load, only a small fraction of its motor units are activated, whereas a greater proportion of the motor units are activated via their motor neurons when the same muscle lifts a heavy load. This mechanism is the major determinant of skeletal muscle performance.
Table 10-1 Mechanisms That Regulate the Contractile Performance of Skeletal and Cardiac Muscle | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In the heart, the number of active muscle fibers cannot be varied because the interiors of adjacent cardiac myocytes are linked by the gap junctions in the intercalated discs (see Chapters 1, 13, and 16). Because these structures provide a low electrical resistance between adjacent cells, the heart operates as a functional syncytium, where depolarization of any region normally activates the entire heart.
Changes in Sarcomere Length
The length-tension relationship allows preload to influence the performance of both skeletal and cardiac muscle (see Chapter 6). In most skeletal muscles, rest length is determined largely by the angles at the joints; as these angles are generally chosen to optimize leverage, rather than to set sarcomere length along the length-tension curve, variations in rest length are of little regulatory importance.
Changes in sarcomere length play a key role in matching the heart’s output during systole to the return of blood during diastole. Because cardiac muscle normally operates on the ascending limb of the length-tension curve (see Chapter 6), an increase in the return of blood during diastole increases the ability of the heart to eject during systole. These length-dependent changes allow the heart to “fine tune” its performance to match venous return and cardiac output, to vary cardiac performance in response to short-term hemodynamic changes, and to equalize the outputs of the right and left ventricles (see below).
Inotropic and Lusitropic Changes
Changes in contractility and relaxation play little role in skeletal muscle, where the high content of sarcoplasmic reticulum allows excitation-contraction coupling to deliver enough calcium to saturate virtually all of the binding sites on troponin C; as a result, even under basal conditions, skeletal muscle tension is at or near the maximum that the muscle can generate. Although trains of stimuli normally maximize tension by generating brief tetani, periods of inactivity allow time for skeletal muscles to relax completely when their tasks are completed.
The situation in the heart, however, is quite different. Because excitation-contraction coupling does not provide enough activator calcium to saturate all of the troponin C in myocardial cells operating under basal conditions, myocardial contractility and relaxation can be regulated by modification of the calcium cycles described in Chapter 7. These functional responses allow the heart to meet a variety of sustained physiological stresses, such as the hemodynamic defense reactions described in Chapter 8. Furthermore, long-term changes in the demands on the heart evoke proliferative responses that change the architecture and composition of the myocardium (see Chapters 9 and 18).
Functional and Proliferative Responses
Cardiac performance is regulated by three fundamentally different types of response; two are functional and the third is proliferative (Table 10-2). Changes in organ physiology represent functional responses that appeared ∼600 million years ago with the evolution of large multicellular animals that required a circulatory system. A second type of functional response, changes in cell biochemistry and biophysics, emerged ∼2,000 million years ago, when the evolution of the intracellular membrane systems allowed single-celled eukaryotes to regulate their internal environment (milieu intárieur). The third type of response, regulation by altered molecular composition, is the most primitive, having appeared at the dawn of life more than 3,500 million years ago. Having had the longest time to evolve, it is not surprising that proliferative responses are the most complex.
The rates at which these responses operate differ. Physiological responses appear most rapidly, often from beat to beat, while biochemical and biophysical responses generally require several seconds or minutes to modify cardiac performance. Proliferative responses, which are responsible for changes in cardiac performance in chronically overloaded and diseased hearts, are brought about by long-term structural modifications in the heart that can take days, weeks, and even months to develop.
Table 10-2 Mechanisms That Regulate Cardiac Performance | |||||
|---|---|---|---|---|---|
|
Physiological responses adjust cardiac performance to meet short-term changes in hemodynamics like an increase in venous return, while biochemical and biophysical responses allow cardiac performance to respond to more sustained hemodynamic challenges like those that occur during exercise. The latter include changes in myocardial contractility and relaxation brought about by modifications of the calcium fluxes between the cytosol, extracellular fluid, and sarcoplasmic reticulum (see Chapter 7). Molecular responses, which depend on proliferative signaling, change the size, shape, and composition of the heart in response to long-term challenges like chronic hemodynamic overloading and disease.
Functional Responses: Physiological and Biochemical Regulation: Changes in End-Diastolic Volume and Myocardial Contractility
The two types of functional responses that control cardiac performance are readily understood by comparing the effects of changing rest length and changing contractility (see Chapter 6). Starling’s law of the heart, an example of physiological regulation, allows changes in end-diastolic volume to match the output of the heart during systole to the return of blood during diastole (Starling, 1918). This mechanism, which had been known to physiologists since the middle of the 19th century (Katz, 2002), was generally viewed as the only determinant of myocardial performance until the mid-1950s, when mechanisms other than changes in end-diastolic volume were shown to regulate contractile performance (see below). The latter, which represent changes in contractility, are initiated by biochemical and biophysical mechanisms that modify the interactions between the heart’s contractile proteins.
Regulation by changing end-diastolic volume and changing contractility were initially thought to arise from entirely different mechanisms. This interpretation was based largely on the now-discarded view that the length-tension relationship in cardiac muscle is caused when changes in the overlap between the thick and thin filaments vary the number of potential interactions between actin and myosin (the “ultrastructural mechanism” described in Chapter 6), whereas variations in contractility are brought about by changes in the chemistry of these interactions. Even though control by changing end-diastolic volume, like that by changing contractility, is now known to result from variations in excitation-contraction coupling, the traditional distinction remains useful because these regulatory mechanisms serve different physiological functions.
Regulation by Changing End-Diastolic Volume
Starling’s law of the heart can be viewed as a mechanism that maintains hemodynamic equilibria by allowing changes in preload and afterload to modify cardiac performance.
Responses to Changing Preload
Increased venous return to the right atrium, for example, when the legs are elevated, causes an increase in right atrial pressure that is transmitted immediately across the tricuspid valve to increase right ventricular end-diastolic volume. According to Starling’s law, the higher preload restores the equilibrium between venous return and right ventricular output by enhancing the
ability of the right ventricle to eject. This ability of the heart to modify its output in response to changes in filling represents a positive feedback in which altered blood flow into the heart leads to a corresponding change in the flow of blood out of the heart.
ability of the right ventricle to eject. This ability of the heart to modify its output in response to changes in filling represents a positive feedback in which altered blood flow into the heart leads to a corresponding change in the flow of blood out of the heart.
Matching the Outputs of the Right and Left Ventricles
Operation on the ascending limb of the length-tension curve (see Chapter 6) provides a positive feedback that plays an essential role in equalizing the outputs of the two ventricles. For example, when leg elevation increases right ventricular output (see above), the increased flow of blood through the lungs increases venous return to the left ventricle which, according to Starling’s law, increases left ventricular ejection. Conversely, reduced ejection by the right ventricle decreases left ventricular filling, and so reduces left ventricular output.
Responses to Changing Afterload
A sudden increase in arterial pressure (afterload), which reduces ventricular ejection (see Chapter 6), increases the volume of blood remaining in the ventricle at the end of systole (end-systolic volume). When the venous return during the next diastole is added to the greater end-systolic volume, end-diastolic volume is increased which, according to Starling’s law, compensates for the reduced ejection by increasing ventricular performance. This leads to an increase in ejection that restores the equilibrium between the amount of blood ejected during systole to the return of venous blood during diastole.
Regulation by Changing Inotropic Properties (Myocardial Contractility or Systolic Function)
Although Starling’s law plays a central role in beat-to-beat and short-term regulation of cardiac performance, length-dependent changes are of limited importance when circulatory changes are sustained. Evidence that factors other than end-diastolic volume regulate cardiac performance is found in Starling’s original papers, which document a positive inotropic effect, sometimes called “homeometric autoregulation” or the “Anrep effect,” which develops after a sudden increase in left ventricular systolic pressure. Once thought to be caused by an undiscovered mechanism within cardiac myocytes, this increase in contractility is now recognized to occur when delayed dilatation of the coronary microcirculation overcomes the negative inotropic effect of the state of energy starvation that occurs when the increased systolic wall stress reduces blood flow to the left ventricle. A more important exception to Starling’s law, described more than 75 years ago, is that the heart can become smaller during upright exercise in humans, even though cardiac output increases several fold (Nylin, 1934). By the mid-1950s, a substantial body of evidence indicated that regulatory mechanisms other than end-diastolic fiber length regulate cardiac performance (Katz et al., 1955), but it was not until Sarnoff described the “family of Starling curves” (1955) that the role of changes in myocardial contractility became clear. Contractility is often referred to as inotropy; a positive inotropic intervention increases contractility while a negative inotropic intervention reduces contractility.
What is Myocardial Contractility?
Myocardial contractility is virtually impossible to define because it is a manifestation of all of the factors, except for preload and afterload (see Chapter 6), that influence the interactions between the contractile proteins. The simplest definition, the ability of the ventricle to do work
at a given preload and afterload, reflects the fact that load-dependent changes in performance do not reflect variations in contractility, but instead are different ways that a muscle operating at a given level of contractility can perform work. Unfortunately, this definition is of limited practical value because it is difficult to quantify preload and afterload in patients. A change in myocardial contractility, however, is easier to define; this is simply any change in cardiac performance that is not caused by altered end-diastolic volume (preload) or arterial pressure (afterload). If, for example, a drug is given to an isolated heart when end-diastolic volume and the resistance to ejection are held constant, and ejection and/or developed pressure increases, then contractility has increased (Fig. 10-1A); conversely, a drug that reduces the ability of this heart to eject or develop pressure has decreased contractility (Fig. 10-1B). Even when it is possible to identify a change in myocardial contractility, there remains the question as to what exactly has been changed. Simple answers fail because contractility is determined by all of the factors, except for load, that influence the ability of a muscle to do work.
at a given preload and afterload, reflects the fact that load-dependent changes in performance do not reflect variations in contractility, but instead are different ways that a muscle operating at a given level of contractility can perform work. Unfortunately, this definition is of limited practical value because it is difficult to quantify preload and afterload in patients. A change in myocardial contractility, however, is easier to define; this is simply any change in cardiac performance that is not caused by altered end-diastolic volume (preload) or arterial pressure (afterload). If, for example, a drug is given to an isolated heart when end-diastolic volume and the resistance to ejection are held constant, and ejection and/or developed pressure increases, then contractility has increased (Fig. 10-1A); conversely, a drug that reduces the ability of this heart to eject or develop pressure has decreased contractility (Fig. 10-1B). Even when it is possible to identify a change in myocardial contractility, there remains the question as to what exactly has been changed. Simple answers fail because contractility is determined by all of the factors, except for load, that influence the ability of a muscle to do work.
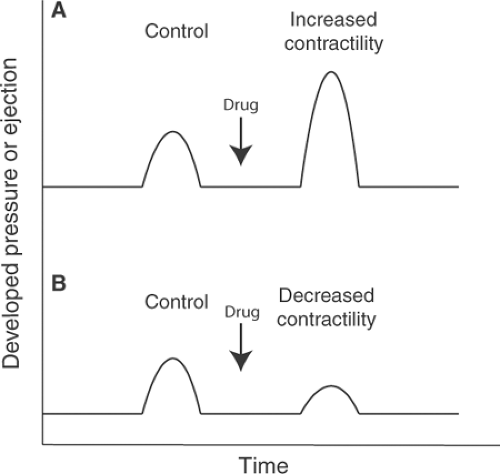 Fig. 10-1: Changing contractility in an isolated heart contracting with constant end-diastolic volume and resistance to ejection. An increase in myocardial contractility (A) increases the developed pressure, volume ejected, or both, while a decrease in myocardial contractility (B) reduces developed pressure, the volume ejected, or both. |
Force-velocity curves were once believed to hold the key to quantifying myocardial contractility, which was thought to be the sole determinant of Vmax (see Chapter 3). This view was based on two assumptions: that changing preload modifies only Po but not Vmax, and that the influence of afterload can be eliminated by extrapolating the curves to zero load where shortening velocity is maximal. However, both assumptions turned out to be incorrect. The first assumption was based on the “ultrastructural mechanism” for the length-tension relationship, which attributed length-dependent variations in the ability of the heart to do work entirely to changes in the number of active cross-bridges. Today, however, several determinants of contractile protein shortening are also known to be modified by changes in sarcomere length (see Chapter 6), which means that altering preload can modify Vmax as well as Po. The second assumption, that sarcomere shortening is the only determinant of Vmax, is also incorrect because of the high resting tension and internal elasticity of cardiac muscle (see Chapter 6) and the spiral arrangement of the fibers in the walls of the heart (see Chapter 1), all of which contribute elasticities that modify both tension and shortening velocity as active state increases and
decreases during each cardiac cycle. Furthermore, those portions of the force-velocity curves that can be measured in beating hearts are not hyperbolic because the active state in cardiac muscle changes throughout systole (see Chapter 6); this means that Po and Vmax cannot be accurately extrapolated in cardiac muscle.
decreases during each cardiac cycle. Furthermore, those portions of the force-velocity curves that can be measured in beating hearts are not hyperbolic because the active state in cardiac muscle changes throughout systole (see Chapter 6); this means that Po and Vmax cannot be accurately extrapolated in cardiac muscle.
Even though efforts to quantify myocardial contractility are of limited accuracy, the concept has considerable clinical significance. This is because any effort to determine what might have happened to a patient who experiences an unexpected change in hemodynamics, such as a fall in blood pressure, requires an understanding of the potential contributions of changes in preload, afterload, and contractility. It is only in this way that a therapeutic regimen most appropriate for the individual patient can be formulated and its effects evaluated.
Regulation by Changes in Lusitropic Properties (Filling or Diastolic Function)
The clinical importance of lusitropic abnormalities was not generally appreciated until the 1970s, almost 20 years after the role of changing contractility had been recognized. This is largely because the initial clinical applications of hemodynamic physiology focused on pressures, which were readily measured by the equipment that was then available. These led to the early use of pressure-based indices, such as the rate of pressure rise during isovolumic contraction (+dP/dt), to quantify myocardial contractility (see Chapter 12). Initial efforts to quantify lusitropic state were also based on pressure measurements; in this case the rate of pressure fall during isovolumic relaxation (-dP/dt). However, the latter is highly dependent on ventricular systolic pressure, so that use of pressure measurements to define lusitropic properties requires corrections that are both difficult and imprecise. It was not until echocardiography and nuclear techniques made it possible to measure changes in the rate and extent of ventricular filling that diastolic function could be characterized in patients (see Chapter 12).
Regulation by Changes in Coronary Perfusion Pressure (The Garden Hose Effect)
The finding that changing coronary perfusion pressure can modify the ability of the heart to develop tension, even after the ventricles are drained of blood and cavity pressure is zero (Salisbury et al., 1960), revealed the existence of an unexpected regulatory mechanism. Evidence that increasing coronary arterial pressure has an “erectile” effect that increases cardiac oxygen consumption (Vogel et al., 1982) demonstrated that the heart’s performance is regulated by distension within its walls, much as occluding the outlet of a garden hose increases its internal diameter. This mechanism, often called the garden hose effect, was initially explained by postulating that higher intramyocardial pressures increase cardiac performance by increasing sarcomere length (Starling’s law of the heart). However, evidence that this phenomenon cannot be explained simply on the basis of changes in sarcomere length (Kitakaze and Marban, 1989; Koretsune et al., 1991; May-Newman et al., 1994; Matsushita et al., 1995) suggests that the garden hose effect occurs when cytoskeletal deformation modifies the intensity of excitation-contraction coupling.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


