Redox Signaling and Oxidative Stress in Lung Diseases
INTRODUCTION
Molecular oxygen is a prerequisite to life of all aerobic organisms. The human lung with its large surface area and extensive blood supply is engineered for its primary function in gas exchange. While oxygen is essential for its many roles in human physiology, high concentrations of oxygen or its metabolites, commonly referred to as reactive oxygen species (ROS), have the potential to cause cellular injury and contribute to disease pathogenesis. The most damaging forms of ROS are free radicals. A free radical, by definition, refers to any chemical species containing one or more unpaired electrons in their atomic or molecular orbitals. These unpaired electron(s) give considerable reactivity to free radical species, which can trigger chemical reactions that damage cellular constituents of living organisms. Molecular oxygen (dioxygen) is itself, a radical based on the presence of unpaired electrons in its outermost orbital; however, their parallel spin retrains its reactivity. O2 can form the superoxide anion radical (![]() ) upon addition of an electron; thus, overcoming this restraint and making
) upon addition of an electron; thus, overcoming this restraint and making ![]() a highly reactive species.1,2 The photodynamic activation of oxygen can result in the formation of singlet oxygen, and its reductive activation results in the formation of hydrogen peroxide (H2O2) or the highly reactive hydroxyl radical (•OH).3,4 When two free radicals share their unpaired electrons, nonradical species of lower reactivity are generated. Thus, ROS constitute both free radicals and nonradicals.
a highly reactive species.1,2 The photodynamic activation of oxygen can result in the formation of singlet oxygen, and its reductive activation results in the formation of hydrogen peroxide (H2O2) or the highly reactive hydroxyl radical (•OH).3,4 When two free radicals share their unpaired electrons, nonradical species of lower reactivity are generated. Thus, ROS constitute both free radicals and nonradicals.
Nitric oxide (NO•) is another small gaseous molecule that serves as an important signaling molecule in diverse physiological processes, including vasorelaxation and immune regulation during chronic inflammation in the lung.5,6 The regulated production of NO• by lung cells is critical for homeostasis of the lung. However, in some contexts, the reaction of NO• with ![]() to form reactive nitrogen species (RNS), such as peroxynitrite (ONOO–), may contribute to the pathophysiology of chronic lung diseases.7–9
to form reactive nitrogen species (RNS), such as peroxynitrite (ONOO–), may contribute to the pathophysiology of chronic lung diseases.7–9
ROS and RNS together play important roles in regulation of cell proliferation, differentiation, and survival.10–12 ROS/RNS can inactivate enzymes including antiproteases, induce apoptosis, regulate cell proliferation, and modulate the immune-inflammatory system in the lungs and other tissues.13–16 ROS/RNS have been implicated in initiating inflammatory responses in the lungs through the activation of transcription factors, protein kinase pathways, chromatin remodeling, and gene expression of proinflammatory mediators.13–16 Under normal physiological conditions, the balance between generation and elimination of ROS/RNS maintains the functional integrity of redox-sensitive signaling cascades regulating cellular phenotypes. In this context, it is important to differentiate the roles of ROS/RNS in “oxidative stress” from “redox signaling.” Paradoxically, it appears that nature has co-opted the chemical reactivities of ROS/RNS to function as signaling molecules in homeostasis and normal cellular physiology.
Redox homeostasis of cells/tissues is maintained by the regulated balance of oxidant production and antioxidant systems, both enzymatic and nonenzymatic. However, an increase in oxidant generation in excess of the capacity of cells/tissues to detoxify or scavenge reactive species leads to oxidative stress.10,17,18 Oxidative stress typically causes damage to cellular components in an indiscriminant manner. Such states are accelerated in the presence of transition metals, such as iron and copper, and/or specific monooxygenases or oxidases.13,15,16 It is also important to recognize that aberrations in redox signaling or oxidative stress may contribute to the pathogenesis of lung diseases. In this chapter, we review the physiology of ROS and RNS; cellular antioxidant systems; and the role of oxidative stress and redox signaling in the development and progression of selected acute and chronic lung diseases.
METABOLISM OF REACTIVE OXYGEN/NITROGEN SPECIES
 ENZYMATIC AND NONENZYMATIC SOURCES OF ROS
ENZYMATIC AND NONENZYMATIC SOURCES OF ROS
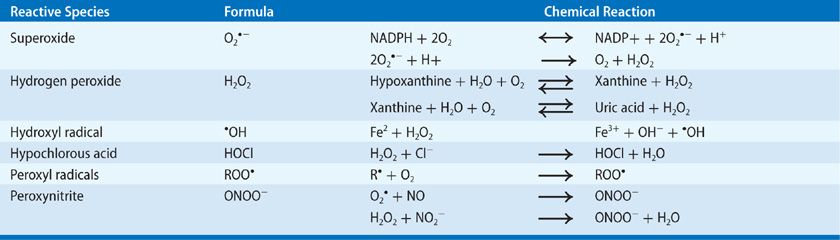
The primary ROS include superoxide anion (![]() ), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), which can be generated from both enzymatic and nonenzymatic sources. The human lung is constantly exposed to ambient air, which may contain environmental toxins capable of inducing ROS generation.13,19 ROS may also be generated by electron transfer reactions in the mitochondria and endoplasmic reticulum (ER), from xenobiotics, and a range of metabolic enzymes that catalyze oxidation reactions.19 The major endogenous ROS/RNS and the primary mechanisms for their generation are summarized in Table 27-1.
), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), which can be generated from both enzymatic and nonenzymatic sources. The human lung is constantly exposed to ambient air, which may contain environmental toxins capable of inducing ROS generation.13,19 ROS may also be generated by electron transfer reactions in the mitochondria and endoplasmic reticulum (ER), from xenobiotics, and a range of metabolic enzymes that catalyze oxidation reactions.19 The major endogenous ROS/RNS and the primary mechanisms for their generation are summarized in Table 27-1.
TABLE 27-1 Key Reactive Oxygen and Nitrogen Species
The highly reactive ![]() formed by the addition of an electron to molecular oxygen is unstable with a half-life of milliseconds. The major site for producing
formed by the addition of an electron to molecular oxygen is unstable with a half-life of milliseconds. The major site for producing ![]() is the mitochondria, which is also the primary site for ATP generation in any cell. During mitochondrial electron transport, there is an estimated leak of 1% to 3% of electrons to oxygen resulting in the formation of
is the mitochondria, which is also the primary site for ATP generation in any cell. During mitochondrial electron transport, there is an estimated leak of 1% to 3% of electrons to oxygen resulting in the formation of ![]() .16
.16 ![]() generated in the mitochondria, due to its negative charge, does not cross membranes and is released into the mitochondrial matrix where it targets proteins with heme moieties or iron–sulfur clusters to cause loss of protein or enzymatic function.20 The steady-state levels of intramitochondrial
generated in the mitochondria, due to its negative charge, does not cross membranes and is released into the mitochondrial matrix where it targets proteins with heme moieties or iron–sulfur clusters to cause loss of protein or enzymatic function.20 The steady-state levels of intramitochondrial ![]() concentrations are maintained at very low levels by detoxifying enzymes. ER is another intracellular compartment in which enzymes that detoxify lipid-soluble drugs and other toxic metabolites, reduce molecular O2 to produce
concentrations are maintained at very low levels by detoxifying enzymes. ER is another intracellular compartment in which enzymes that detoxify lipid-soluble drugs and other toxic metabolites, reduce molecular O2 to produce ![]() and/or H2O2, while oxidizing unsaturated fatty acids and xenobiotics.19,20
and/or H2O2, while oxidizing unsaturated fatty acids and xenobiotics.19,20
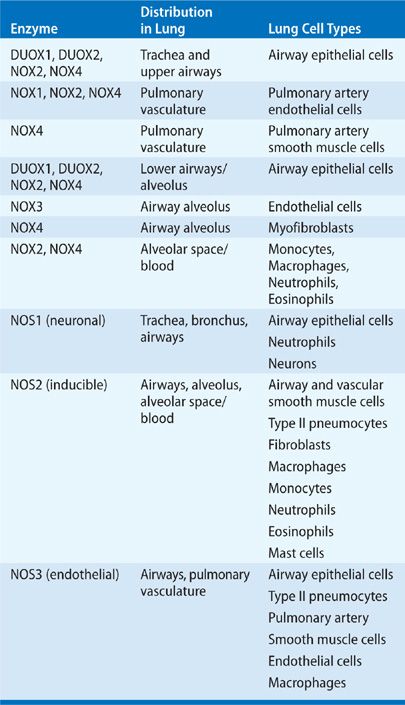
The NADPH oxidase (NOX) gene family encompasses enzymes, the primary function of which is the regulated generation of ROS. NOX enzymes form a membrane-bound multicomponent complex that is present in phagocytes and nonphagocytic cells in the lung.15,16,18 NOX enzymes play crucial roles in host defense, signal transduction, and hormone synthesis in eukaryotes. There are seven mammalian NOX homologs comprising NOX1 to 5 and the DUOX1 and 2.21–24 The best characterized NOX isoform is NOX2, which is essential for microbicidal-killing activity of phagocytes. NOX2, upon activation, generates ROS by coordinated assembly and activation of the NOX2 enzymatic complex, which comprises the membrane-associated flavocytochrome b558 (gp91phox), p22phox, and the various cytosolic cofactors (p47phox, p67phox, and p40phox, and the GTPase, Rac1). This complex then mediates the transmembrane electron transfer from the major electron donor, NADPH, to reduce molecular O2 to ![]() and H2O2.15,16,18
and H2O2.15,16,18
Similar to NOX2, activation of NOX1 and NOX3 also requires association with p22phox, and assembly with Rac1 and cytosolic cofactors (p47phox and p67phox or their homologs, NOX organizer 1 (NOXO1) and NOX activator 1 (NOXA1). NOX4 also requires p22phox, but is constitutively active and functions independently of activation of other cofactors.16,21–24 Expression of NOX1 and NOX4 in nonphagocytic fibroblasts results in increased levels of both ![]() and H2O2, implying that these, unlike the phagocytic NOX2, possess intrinsic basal activity. NOX5 and DUOX1/2 differ from the other NOX homologs and contain additional intracellular Ca2+-binding EF-hand domain regions, and are regulated by Ca2+ signaling, independent of p22phox or other cytosolic factors.16,21–24 The dual oxidases, DUOXs, are composed of the NOX-like region at the C-terminal half, two EF-hands, a membrane-spanning region, and a peroxidase-like domain at the N-terminus. DUOXs do not require cytosolic regulatory components for their activity.16,21–24 However, transmembrane maturation factors, DUOXA1 and DUOXA2, are essential for ER-to-Golgi transition, maturation, and targeting DUOXs to the plasma membrane as functional complexes. Although initially identified in the thyroid gland, DUOX1 and DUOX2 are the primary sources of H2O2 production in the airway epithelium.25 The NOX/DUOX isoforms are expressed widely in the lung extending from the proximal trachea and large airways to terminal bronchioles and alveoli (Table 27-2).16,21–25
and H2O2, implying that these, unlike the phagocytic NOX2, possess intrinsic basal activity. NOX5 and DUOX1/2 differ from the other NOX homologs and contain additional intracellular Ca2+-binding EF-hand domain regions, and are regulated by Ca2+ signaling, independent of p22phox or other cytosolic factors.16,21–24 The dual oxidases, DUOXs, are composed of the NOX-like region at the C-terminal half, two EF-hands, a membrane-spanning region, and a peroxidase-like domain at the N-terminus. DUOXs do not require cytosolic regulatory components for their activity.16,21–24 However, transmembrane maturation factors, DUOXA1 and DUOXA2, are essential for ER-to-Golgi transition, maturation, and targeting DUOXs to the plasma membrane as functional complexes. Although initially identified in the thyroid gland, DUOX1 and DUOX2 are the primary sources of H2O2 production in the airway epithelium.25 The NOX/DUOX isoforms are expressed widely in the lung extending from the proximal trachea and large airways to terminal bronchioles and alveoli (Table 27-2).16,21–25
TABLE 27-2 Enzymatic Sources for ROS and RNS
In biological systems, dismutation reaction of ![]() generates H2O2, either spontaneously or by enzymatic catalysis by superoxide dismutases (SODs).
generates H2O2, either spontaneously or by enzymatic catalysis by superoxide dismutases (SODs). ![]() can be generated by molybdenum hydroxylase reactions involving xanthine, sulfite, and aldehyde oxidases, dihydroorotate and flavoprotein dehydrogenases, tryptophan dioxygenases as well as arachidonic acid metabolism.26,27 Certain oxidases such as monoamine and amino acid oxidases can generate H2O2 directly without the intermediate formation of
can be generated by molybdenum hydroxylase reactions involving xanthine, sulfite, and aldehyde oxidases, dihydroorotate and flavoprotein dehydrogenases, tryptophan dioxygenases as well as arachidonic acid metabolism.26,27 Certain oxidases such as monoamine and amino acid oxidases can generate H2O2 directly without the intermediate formation of ![]() .17 During chronic inflammation in the lung, H2O2 is produced by both resident and inflammatory cells of the lung.26,27 The oxidizing potential of H2O2 is amplified by inflammatory cell peroxidases such as myeloperoxidase (MPO) and/or eosinophil peroxidase (EPO).28–32 The hydroxyl radical, •OH, is the neutral form of the hydroxide ion that is highly reactive toward cellular constituents, more so than
.17 During chronic inflammation in the lung, H2O2 is produced by both resident and inflammatory cells of the lung.26,27 The oxidizing potential of H2O2 is amplified by inflammatory cell peroxidases such as myeloperoxidase (MPO) and/or eosinophil peroxidase (EPO).28–32 The hydroxyl radical, •OH, is the neutral form of the hydroxide ion that is highly reactive toward cellular constituents, more so than ![]() or H2O2. This radical contributes to most of the reactivity by
or H2O2. This radical contributes to most of the reactivity by ![]() and H2O2 in a series of reactions that are catalyzed by transition metal ions. Excess
and H2O2 in a series of reactions that are catalyzed by transition metal ions. Excess ![]() targets Fe–S cluster-containing enzymes and catalyzes production of •OH from H2O2 by reducing free iron (Fe3+ to Fe2+) for Fenton chemistry (see Table 27-1). MPO and EPO enzymes represent an alternate pathway for •OH formation in vivo. MPO uses Cl– as substrate to generate hypochlorous acid, whereas EPO uses Br– to generate brominating species. The hypohalous acids then generate •OH by reaction with
targets Fe–S cluster-containing enzymes and catalyzes production of •OH from H2O2 by reducing free iron (Fe3+ to Fe2+) for Fenton chemistry (see Table 27-1). MPO and EPO enzymes represent an alternate pathway for •OH formation in vivo. MPO uses Cl– as substrate to generate hypochlorous acid, whereas EPO uses Br– to generate brominating species. The hypohalous acids then generate •OH by reaction with ![]() .33–38 Both these enzymes can accelerate oxidative modifications of proteins, in particular bromination and chlorination.
.33–38 Both these enzymes can accelerate oxidative modifications of proteins, in particular bromination and chlorination.
 ENZYMATIC AND NONENZYMATIC SOURCES OF RNS
ENZYMATIC AND NONENZYMATIC SOURCES OF RNS
An important RNS in the lung is NO•, which is endogenously generated by specific nitric oxide synthases (NOSs). These NOS enzymes metabolize L-arginine to NO• and L-citrulline via a five electron oxidation reaction.6,39–48 This reaction requires a dimeric enzyme, oxygen, NADPH and the cofactors, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), tetrahydrobiopterin (BH4), calmodulin, and iron protoporphyrin. Active NOS enzymes exist in the dimeric form with each monomer consisting of an N-terminal oxygenase domain that binds heme, BH4, and substrate L-arginine. The cofactors FAD, FMN, and NADPH bind the C-terminal end of the NOS monomer. There are three different NOS forms, all of which are expressed in the lung, the inducible form iNOS or NOS2, neuronal NOS or NOS1, and the endothelial NOS enzyme NOS3 (Table 27-2). The NOS1 and NOS3 enzymes are calcium dependent and produce picomolar levels of NO, while nanomolar levels are generated by the calcium-independent iNOS. NO• synthesis by iNOS is regulated by the availability of the substrate L-arginine and cofactor BH4. Transfer of electrons occur from the carboxy-reductase domain to the heme iron of the oxygenase domain, which then binds oxygen and oxidizes L-arginine to generate the end products, NO• and citrulline.6,39–48 Uncoupling of NOS enzymes can contribute to the formation of the ![]() . In a low arginine condition, NADPH is oxidized by the enzyme to generate
. In a low arginine condition, NADPH is oxidized by the enzyme to generate ![]() . Arginase, an enzyme that competes with the NOSs for L-arginine decreases the arginine availability for NOS. Arginase is a critical enzyme in the urea cycle and converts arginine to ornithine, and then to urea; it can promote uncoupling of NOSs to generate
. Arginase, an enzyme that competes with the NOSs for L-arginine decreases the arginine availability for NOS. Arginase is a critical enzyme in the urea cycle and converts arginine to ornithine, and then to urea; it can promote uncoupling of NOSs to generate ![]() .49,50 About 40% of the highly reactive and highly diffusible NO• is consumed in chemical reactions and, when metabolized, give rise to reactive intermediates. NO• reacts with O2 yielding nitrite (NO2–) and recycling of nitrite causes regeneration of bioactive NO•. NO2– is also a substrate for the heme peroxidases, MPO and EPO, which oxidize nitrite to nitrogen dioxide radical (NO2•). NO• is also oxidized to methemoglobin and NO3– by reaction with oxyhemoglobin. NO• reacts with
.49,50 About 40% of the highly reactive and highly diffusible NO• is consumed in chemical reactions and, when metabolized, give rise to reactive intermediates. NO• reacts with O2 yielding nitrite (NO2–) and recycling of nitrite causes regeneration of bioactive NO•. NO2– is also a substrate for the heme peroxidases, MPO and EPO, which oxidize nitrite to nitrogen dioxide radical (NO2•). NO• is also oxidized to methemoglobin and NO3– by reaction with oxyhemoglobin. NO• reacts with ![]() to form peroxynitrite (ONOO–), which can mediate tyrosine nitration that may result in either the loss or gain of function of proteins.7–9,51 In acidic environments, the protonation of ONOO– results in the formation of ONOOH (peroxynitrous acid), which is then decomposed to both NO3– via intermediate •OH and NO2.52 ONOOH can also react with thiol residues to form S-nitrosothiols (SNOs) and the reaction is referred to as S-nitrosation or S-nitrosylation. A wide variety of proteins including kinases, channels, and transcription factors are susceptible to S-nitrosylation. SNOs are important molecules in the signaling of NO• bioactivity in the respiratory system.53–57 SNOs are present in the airway epithelial lining fluid in μmol concentrations, can influence airway tone, and possess substantially greater half-lives than NO• in the lung. It has also been reported that iNOS can specifically bind to cyclooxgenase-2 (COX-2) and S-nitrosylate COX-2, upregulate its catalytic activity and enhance prostaglandin E2 production. Thus, S-nitrosylation represents an important signaling pathway for NO•.53–57 Under physiological conditions, interaction with metal centers of enzymes and S-nitrosylation are the major mechanisms for biological actions of NO•. In biological systems, the levels of
to form peroxynitrite (ONOO–), which can mediate tyrosine nitration that may result in either the loss or gain of function of proteins.7–9,51 In acidic environments, the protonation of ONOO– results in the formation of ONOOH (peroxynitrous acid), which is then decomposed to both NO3– via intermediate •OH and NO2.52 ONOOH can also react with thiol residues to form S-nitrosothiols (SNOs) and the reaction is referred to as S-nitrosation or S-nitrosylation. A wide variety of proteins including kinases, channels, and transcription factors are susceptible to S-nitrosylation. SNOs are important molecules in the signaling of NO• bioactivity in the respiratory system.53–57 SNOs are present in the airway epithelial lining fluid in μmol concentrations, can influence airway tone, and possess substantially greater half-lives than NO• in the lung. It has also been reported that iNOS can specifically bind to cyclooxgenase-2 (COX-2) and S-nitrosylate COX-2, upregulate its catalytic activity and enhance prostaglandin E2 production. Thus, S-nitrosylation represents an important signaling pathway for NO•.53–57 Under physiological conditions, interaction with metal centers of enzymes and S-nitrosylation are the major mechanisms for biological actions of NO•. In biological systems, the levels of ![]() are 10–11 to 10–10 M while that of NO is 10–9 to 10–7 M. Under these conditions, ONOO– is formed at a low rate by the reaction of NO• with
are 10–11 to 10–10 M while that of NO is 10–9 to 10–7 M. Under these conditions, ONOO– is formed at a low rate by the reaction of NO• with ![]() with a 1:1 stoichiometry.7–9,51,53–57 However, under pathophysiological conditions, ONOO– and its derivatives are formed at high levels and may cause irreversible damage to the respiratory chain, inhibit ATP synthesis, and induce cytochrome c release and caspase-dependent apoptosis. RNS may also mediate lipid peroxidation, protein oxidation and nitration, enzyme inactivation, or even cell necrosis.7–9,51,53–57
with a 1:1 stoichiometry.7–9,51,53–57 However, under pathophysiological conditions, ONOO– and its derivatives are formed at high levels and may cause irreversible damage to the respiratory chain, inhibit ATP synthesis, and induce cytochrome c release and caspase-dependent apoptosis. RNS may also mediate lipid peroxidation, protein oxidation and nitration, enzyme inactivation, or even cell necrosis.7–9,51,53–57
MECHANISMS TO DETOXIFY ROS/RNS
ROS/RNS play a key role in the initiation, amplification, and persistence of inflammation associated with chronic lung diseases.15–18,58,59 Similar to the formation of ROS/RNS, the removal or detoxification of these species may involve enzymatic and nonenzymatic mechanisms. Nonenzymatic systems constitute the first line of defense against ROS/RNS. The low–molecular-weight nonenzymatic antioxidants in the lung include glutathione, vitamins C and E, β-carotene, uric acid, thiols, and taurine. The larger–molecular-weight antioxidants include lactoferrin, albumin, ceruloplasmin, and transferrin; these molecules mediate their antioxidant function by binding heavy metals, making them unavailable for participation in the Fenton reaction to produce free radicals such as hydroxyl radical.15,18,26,59,60 Nonenzymatic antioxidant activities range from the hydrophilic quenching of free radicals, protection of critical sulfhydryl groups on proteins to inhibition of lipid peroxidation.
Although nonenzymatic antioxidants are the first line of defense against different ROS, several enzymatic antioxidants work in concert with nonenzymatic antioxidants to form a tightly regulated antioxidant network. The major components of the pulmonary enzymatic antioxidant defense systems are SODs, catalase, and glutathione peroxidases. Peroxiredoxins (PRXs), thioredoxins (TRXs), glutaredoxins, heme oxygenases, and reductases are also involved in cellular adaptation and protection against oxidative stress in the lung. The enzymatic reactions that catalyze the elimination of ROS/RNS are summarized in Figure 27-1.
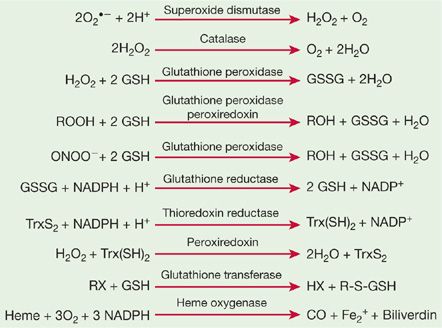
Figure 27-1 Enzymatic mechanisms to detoxify reactive oxygen and nitrogen species.
Superoxide Dismutases: The ![]() radical is a primary oxidant in chronic lung inflammation.15,17,61 It participates in the generation of other reactive metabolites, H2O2, •OH, and ONOO–. SODs represent a key defense against reactive species generated during normal metabolism or inflammatory states. SODs are ubiquitous enzymes that catalyze the dismutation of
radical is a primary oxidant in chronic lung inflammation.15,17,61 It participates in the generation of other reactive metabolites, H2O2, •OH, and ONOO–. SODs represent a key defense against reactive species generated during normal metabolism or inflammatory states. SODs are ubiquitous enzymes that catalyze the dismutation of ![]() radical to the weaker oxidant, H2O2 (see Fig. 27-1). The function of SOD extends beyond its catalysis of
radical to the weaker oxidant, H2O2 (see Fig. 27-1). The function of SOD extends beyond its catalysis of ![]() dismutation to its participation in the regulation of normal cellular homeostasis. H2O2 formed from the SOD-catalyzed reaction may function as a signaling molecule at low concentrations. By reducing steady-state concentrations of
dismutation to its participation in the regulation of normal cellular homeostasis. H2O2 formed from the SOD-catalyzed reaction may function as a signaling molecule at low concentrations. By reducing steady-state concentrations of ![]() , which reacts with NO• to form ONOO–, SOD may serve to promote vasoreactivity and reduce inflammation.15,61
, which reacts with NO• to form ONOO–, SOD may serve to promote vasoreactivity and reduce inflammation.15,61
There are three mammalian SOD isozymes: the intracellular copper-zinc SOD (CuZn-SOD), the mitochondrial manganese SOD (Mn-SOD), and extracellular SOD (EC-SOD).61 The structural and catalytic characteristics of these SOD isozymes are highlighted in Table 27-3.62–65 Given the complexity of the human lung with its plethora of different cell types, it is conceivable that these isozymes are expressed differentially in specific lung cells with subcellular compartmentalization, as summarized in Table 27-4.66–79
TABLE 27-3 Characteristics of Superoxide Dismutases
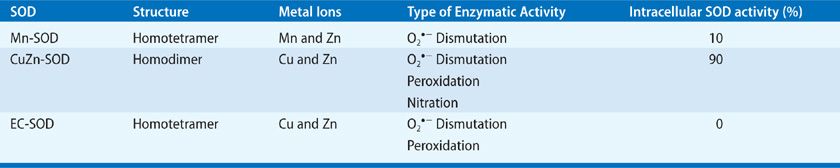
TABLE 27-4 Enzymatic Mechanisms to Detoxify or Remove ROS/RNS
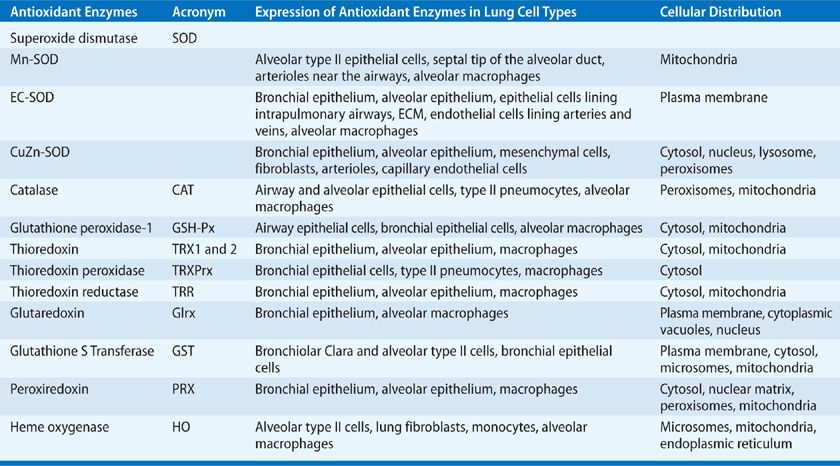
Aside from dismutating ![]() radical by the cyclic oxidation–reduction of its metal ion Cu2+, CuZn-SOD exhibits peroxidase activity.65 At high levels, H2O2 may reduce the Cu2+ to either Cu+-O or Cu2+-OH that may oxidize the histidine residue in the monomer and inactivate or oxidize lung proteins.80–82 CuZn-SOD also nitrates tyrosine residues in proteins via peroxynitrite,83–87 and catalyzes the release of NO from nitrosothiols.88 Although Mn-SOD has similar dismutase activity, it does not elicit peroxidation or nitration, and is inactivated by tyrosine nitration, but not by hydrogen peroxide or cyanide.
radical by the cyclic oxidation–reduction of its metal ion Cu2+, CuZn-SOD exhibits peroxidase activity.65 At high levels, H2O2 may reduce the Cu2+ to either Cu+-O or Cu2+-OH that may oxidize the histidine residue in the monomer and inactivate or oxidize lung proteins.80–82 CuZn-SOD also nitrates tyrosine residues in proteins via peroxynitrite,83–87 and catalyzes the release of NO from nitrosothiols.88 Although Mn-SOD has similar dismutase activity, it does not elicit peroxidation or nitration, and is inactivated by tyrosine nitration, but not by hydrogen peroxide or cyanide.
EC-SOD is the primary extracellular SOD in the lung72 containing Cu and Zn ions.89,90 The EC-SOD has an active site inhibitable by H2O2 and cyanide, similar to CuZn-SOD.74–76 EC-SOD has an arginine- and lysine-rich heparin/matrix-binding domain at its C-terminal region.91–94 Its extracellular localization is maintained by the interaction of this domain with heparin and heparin sulfate proteoglycans on diverse lung cell types and in the extracellular matrix (ECM) of the lung. This heparin/matrix-binding polybasic region is sensitive to proteolysis, leading to reduced affinity of EC-SOD for ECM and increased release of EC-SOD in plasma.72,91–97 EC-SOD is abundant in the lung both at the mRNA and protein levels.91–94 As indicated in Table 27-4, the wide cellular distribution of EC-SOD is consistent with robust EC-SOD activity in the human lung (eightfold > liver, sixfold > brain, two- to threefold > heart, and one- to twofold > kidney)61; in comparison, CuZn-SOD and Mn-SOD activities are much lower. The lower activities of CuZn-SOD and Mn-SOD in the lung suggest that cytoplasmic production of ![]() is lower in lung cells, compared with the metabolically more active cells of the liver and kidney. The dense airway and vascular network in the lung and the higher potential for extracellular inflammatory events resulting from direct exposure of the lung to the external environment may account for high EC-SOD levels in the lung. Even with its high lung localization, EC-SOD is not present at sufficient concentrations to justify its role as the sole scavenger of
is lower in lung cells, compared with the metabolically more active cells of the liver and kidney. The dense airway and vascular network in the lung and the higher potential for extracellular inflammatory events resulting from direct exposure of the lung to the external environment may account for high EC-SOD levels in the lung. Even with its high lung localization, EC-SOD is not present at sufficient concentrations to justify its role as the sole scavenger of ![]() across the entire extracellular space. While CuZn-SOD and Mn-SOD function mainly as bulk scavengers of the
across the entire extracellular space. While CuZn-SOD and Mn-SOD function mainly as bulk scavengers of the ![]() radicals, the relatively high levels of EC-SOD in the lung and its binding specificity to the ECM components provide protection for the lung matrix.14,98
radicals, the relatively high levels of EC-SOD in the lung and its binding specificity to the ECM components provide protection for the lung matrix.14,98
Lack of abnormalities in CuZn-SOD–deficient mice suggest that pathological consequences of mutations are perhaps due to gain of compensatory functions of the enzyme including its peroxidase or nitration reactions and not associated just with the complete loss of SOD activity.97,99 EC-SOD–overexpressing mice were only partially protected from hyperoxia-induced lung injury, influenza, bleomycin, and hemorrhagic shock,14,61,98 suggesting that EC-SOD may not be sufficient to attenuate oxidative stress in pathological states in which the EC-SOD system is overwhelmed. Under hyperoxia, mice lacking EC-SOD show shortened survival and extensive lung damage.14,61,98 Thus, although other antioxidant enzymes may compensate for the loss of EC-SOD under homeostatic conditions, during inflammatory stress, EC-SOD is essential for protecting the lung and limiting injury. Over 90 genetic polymorphisms have been identified in CuZn-SOD, several in association with neurodegenerative diseases.62 The Arg 213-gly polymorphism in EC-SOD (R213G) is found in 4% to 6% of the human population and influences chronic obstructive pulmonary disease100,101 and acute lung injury.102
Catalase: H2O2 is reduced to water by catalase (CAT) and the glutathione peroxidases (see Fig. 27-1). CAT is a metalloprotein oxidoreductase enzyme widely expressed in lung cells (see Table 27-4).62,103–106 In the presence of excess H2O2, CAT undergoes alternate divalent oxidation and reduction at its heme-containing active site. Catalase degrades H2O2 to O2 and water.62,103–106 Although catalase is the principal scavenger of H2O2, it is unable to metabolize large molecular peroxides including lipid peroxides. The gene for catalase is not induced by oxidant stress. However, posttranslational tyrosine phosphorylation of CAT upregulates its activity107; whereas, oxidation of tyrosine residues inhibits CAT activity.8
Glutathione peroxidases: Glutathione peroxidases (GSH-Pxs) are selenocysteine-containing tetrameric enzymes that utilize reduced glutathione (GSH), a low–molecular-weight tripeptide, as an electron donor and catalyze the biotransformation of various organic and inorganic peroxides, including H2O2 and lipid peroxides, to their corresponding alcohols. The detoxification of peroxides by GSH-Pxs occurs via bidirectional second-order kinetics and is a saturation-limited process.26,108 GSH-Px1, GSH-Px2, GSH-Px3, and GSH-Px4 are the four glutathione peroxidases.26,108 GSH-Px1 is a ubiquitous intracellular form and is the predominant isoform that catalyzes the removal of inorganic peroxides, lipid peroxides and hydroperoxides.26,108–111 GSH-Px2 is localized to the gastrointestinal epithelia with substrate specificities similar to that of GSH-Px1. GSH-Px3 is a secreted form able to reduce lipid hydroperoxides.110,111 This extracellular isoform accounts for 57% of the GSH-Px activity in the epithelial lining fluid, and GSH-Px1 contributes to 40% activity.110,111 The fourth isoform, GSH-Px4, is an intracellular peroxidase that preferentially catalyzes the peroxidation of phospholipid hydroperoxides.109
Thioredoxins: Thioredoxins (TRXs), which contain an active site cysteine, serve as redox sensors while also reducing H2O2.112,113 H2O2 oxidizes the reduced dithiol group (-SH HS-) in TRX to a disulfide bridge (-S-S-). TRXs can reduce protein disulfides (-SH) and protein sulfenic acid intermediates (-SO3H) by cysteine thiol–disulfide exchanges.10,11 Two human TRXs are expressed widely in various lung cell types (see Table 27-4). In addition to its direct antioxidant function, TRX, in cooperation with PRXs, augments gene expression of other antioxidant enzymes, including SOD.114–118 TRXs also participate in refolding of oxidized proteins and activate transcription factors by reducing cysteines present in the DNA-binding site.119 TRX can be activated by hypoxia, lipopolysaccharide, H2O2, microbial infections, and photochemicals. Thus, TRXs are powerful redox modulators, protects cells against oxidative stress, and participates in cell proliferation and survival.114–118
Glutaredoxins: Glutaredoxins (GRXs) are thiol–disulfide oxidoreductases with antioxidant capacity in human lung (see Table 27-4).18,120–123 GRX regulates cellular redox state and redox-dependent signaling pathways via modulation of protein glutathionylation; it regulates the intracellular and extracellular homeostases of glutathionylated proteins and GSH.18,120–124 These enzymes use glutathione as a cofactor and catalyze the reversible exchange of glutathione with protein thiol groups (see chemical reactions in Fig. 27-1). GRX enzymes are dependent on GSH/GSSG concentrations.125
Glutathione-S-Transferases: Glutathione-S-transferases (GSTs) are detoxification enzymes that require intracellular GSH for their catalytic activity. These antioxidant enzymes inactivate secondary metabolites, such as unsaturated aldehydes, epoxides, and hydroperoxides.126 Three major families of GSTs have been described; the cytosolic GST, mitochondrial GST, and membrane-associated microsomal GST (see Table 27-4).60 GSTs regulate eicosanoid and glutathione metabolism.60,127 Under conditions of oxidative stress, cytosolic GST interacts with PRXs.126 GST family enzymes are expressed in normal lung, mainly in the airways. They protect cells against a number of oxidizing species. GSTs have high genetic variability, and are implicated in the development of smoking-related nonmalignant and malignant diseases.60,128–131
Peroxiredoxins: Peroxiredoxins (PRXs) are broad-spectrum peroxidases that detoxify or reduce H2O2, peroxynitrite, and organic hydroperoxides (ROOH).132,133 These are nonseleno-peroxidases whose antioxidant properties are dependent on redox-active cysteines.133 Six different PRXs have been found in human lung (see Table 27-4).134,135 These PRXs differ widely in their specificities for H2O2, lipid and phospholipid hydroperoxides.134–138 PRX V and VI function as peroxynitrite reductases, and are protective in ROS/RNS-mediated lung injury.134–136,139,140 PRXs also regulate peroxide-mediated signaling cascades related to cell proliferation, differentiation, and apoptosis by modulating cytokine-mediated induction of H2O2.18,133,137,138
Heme oxygenases: Heme oxygenase (HO) catalyzes the breakdown of pro-oxidant heme to generate equimolar amounts of carbon monoxide, ferrous iron, and biliverdin (Fig. 27-1). Biliverdin is then converted by biliverdin reductase to bilirubin, the antioxidant endproduct of the HO reaction.141–144 CO transported to the lung has vasodilatory and antiapoptotic properties.145–150 Iron is used for heme synthesis in the cells required for heme-containing proteins or transported to the bone marrow and other tissues.18,141,144,151,152 There are three isoforms of HO, the inducible HO-1, and the constitutive forms, HO-2 and HO-3. During oxidative stress, transcriptional activation results in rapid induction of HO-1.141,144,145 This adaptive response of HO-1 confers protection during inflammation and oxidative stress. HO-1 is expressed widely in the lung (see Table 27-4). Consistent with the antioxidant properties of HO-1, mice deficient in HO-1 are more susceptible to oxidative stress.153,154 Overexpression or induction of HO-1 suppresses inflammation in several models of chronic lung disease.18,141,144,151,152
CELLULAR SOURCES AND REGULATION OF ROS/RNS
 IMMUNE CELLS
IMMUNE CELLS
Inflammation is an adaptive response to infectious and noninfectious tissue injury. Recruited inflammatory cells emigrate from the pulmonary microcirculation into the airspaces where they become activated to generate ROS/RNS.
NO• participates in pathogen killing by macrophages.155 NO• also delays fusion of phagosomes with lysosomes to form a functional phagolysosome, which enhances antigen processing/presentation of macrophages. Macrophages scavenge endogenous dying cells. Phagocytosis of dying cells requires the secretion of alarmins by dying cells to attract and preactivate phagocytes; these signals by dying cells ensures specific recognition and phagocytosis/efferocytosis, all of which involve redox regulation.156,157
Beyond the chemical interactions of NO• and ROS that cooperatively eradicate pathogens, these redox-active biomolecules regulate cellular metabolism, inflammation, and tissue-repair functions. Cellular supply of substrates/cofactors for iNOS activity, including arginine, is required for NO• production. However, in the absence of arginine or BH4, uncoupled NOS becomes an ![]() /H2O2 generator. The flavin-binding sites of the reductase domain of iNOS, eNOS, and nNOS are a source of
/H2O2 generator. The flavin-binding sites of the reductase domain of iNOS, eNOS, and nNOS are a source of ![]() generation in the absence of arginine.155,158–161 Therefore, metabolic pathways that control arginine and BH4 play a role in determining NO•–
generation in the absence of arginine.155,158–161 Therefore, metabolic pathways that control arginine and BH4 play a role in determining NO•–![]() balance. Cellular arginine levels are dependent on uptake and transport mechanisms and the activation of NOS-arginase enzymatic systems that use arginine. Arginase activation produces ornithine, a starting metabolite for the production of polyamines that are critical molecules supporting DNA stabilization, ion channel transport, and cell proliferation.155,158–161 Arginase is regulated by NOS and NOX activities; N-hydroxyarginine, a product of NOS, inhibits arginase, while
balance. Cellular arginine levels are dependent on uptake and transport mechanisms and the activation of NOS-arginase enzymatic systems that use arginine. Arginase activation produces ornithine, a starting metabolite for the production of polyamines that are critical molecules supporting DNA stabilization, ion channel transport, and cell proliferation.155,158–161 Arginase is regulated by NOS and NOX activities; N-hydroxyarginine, a product of NOS, inhibits arginase, while ![]() increases arginase activity.155,158–161 High arginase activity is associated with elevated ROS and low NO• fluxes. NO• antagonizes NOX2 assembly through the activation of PPARγ which, in turn, inhibits expression of the p47 subunit required for NOX2 activation.155,158–161 Thus,
increases arginase activity.155,158–161 High arginase activity is associated with elevated ROS and low NO• fluxes. NO• antagonizes NOX2 assembly through the activation of PPARγ which, in turn, inhibits expression of the p47 subunit required for NOX2 activation.155,158–161 Thus, ![]() production is suppressed when NOS activity and NO• levels are high. NO• also inhibits COX2 activity, reducing COX2-dependent ROS production.162 Thus, as NO• levels decline, ROS generation may increase via multiple mechanisms.
production is suppressed when NOS activity and NO• levels are high. NO• also inhibits COX2 activity, reducing COX2-dependent ROS production.162 Thus, as NO• levels decline, ROS generation may increase via multiple mechanisms.
The balance between NO• and ROS may play a key role in the orchestration and resolution of inflammation. RNS and ROS actively control innate and adaptive immune signaling. This aspect of redox function is evident in cells of myeloid lineage, such as monocytes, macrophages, and neutrophils. RNS and ROS produced by these cells participate in induction, maintenance, and/or termination of proinflammatory and anti-inflammatory signaling. Similar to the effect of NO• on pathogen eradication, the temporal and spatial concentration profiles of NO• are key determinants of immune-mediated processes.155,158–161 A relationship between increasing steady-state levels of NO• has been linked to regulation of expression of tumor suppressor gene p53 and apoptosis in murine and human macrophage cell lines.163,164 Concentration- and time-dependent changes in the functional profiles of NO• are evident from NO-mediated regulation of cell survival protein signaling cascade165–169
Macrophages rely primarily on the NOX2 complex to produce ROS,5,17,157,168 although the oxidative burst of macrophages is less intense than that of neutrophils. Induction of the proinflammatory phase of an innate immune response is an early response in the immune activation process. This is defined as “classical activation” in macrophages (also called M1 macrophages), and is associated with the production and release of proinflammatory cytokines, proteases including MMP-9, transcription factors such as NF-κB as well as RNS and ROS, including NO• and ![]() . In addition to a role in pathogen eradication, the localized levels of ROS/RNS may dictate integrated signaling and the type of immune activation and determination of cellular phenotypes; ROS/RNS may also regulate crosstalk between proinflammatory and resolution pathways. Classical (proinflammatory) activation of macrophages is followed by an anti-inflammatory healing/tissue-repair phase, which is the ideal outcome of a successful innate immune response. These phases of the innate immune response are initiated by the pathogens or tissue injury, but are rapidly reenforced by the actions of anti-inflammatory cytokines released from macrophages that function in an autocrine manner.157,158,170–172 These cytokines initiate downregulation of the proinflammatory phase and induction of the repair or tissue remodeling phase.
. In addition to a role in pathogen eradication, the localized levels of ROS/RNS may dictate integrated signaling and the type of immune activation and determination of cellular phenotypes; ROS/RNS may also regulate crosstalk between proinflammatory and resolution pathways. Classical (proinflammatory) activation of macrophages is followed by an anti-inflammatory healing/tissue-repair phase, which is the ideal outcome of a successful innate immune response. These phases of the innate immune response are initiated by the pathogens or tissue injury, but are rapidly reenforced by the actions of anti-inflammatory cytokines released from macrophages that function in an autocrine manner.157,158,170–172 These cytokines initiate downregulation of the proinflammatory phase and induction of the repair or tissue remodeling phase.
NO• also affects function of T lymphocytes.155,173–176 Low NO• concentrations promote differentiation of IFN-γ producing Th1 (T helper 1) in mice and humans, mediated by cGMP activation.177 During the immune repair/restoration phase, the collective activity of IL-4, IL-13, IL-10, and TGF-β suppress iNOS expression, thereby decreasing NO• and shifting in favor of ROS.155,173–177 An NO•-independent role for arginase has been identified in the differentiation of alternatively activated macrophages. Although arginase expression in macrophages is prominently associated with Th2 responses, part of its function is to sequester arginine away from effector T cells resulting in a reduced Th2 response; reconstitution with exogenous arginine blocks this reduced Th2 response. In general, low NO• favors Th2 responses, and high NO• augments Th1 responses, suggesting that a NO•/ROS balance may be a critical determinant of immune polarity.155,178,179 Naïve lymphocytes exposed to high μmol concentrations of NO•, however, regulate the expansion and proliferation of regulatory T cells within lymphoid tissue.155
This concentration-dependent regulation by NO• is seen in immunosuppression by heterogeneous immature myeloid cells called myeloid-derived suppressor cells (MDSCs).180,181 Free radical producing subsets of these MDSCs are critical regulators of allergic airway inflammation.182,183 NO•-producing cells suppress T cell proliferation and airway hyperresponsiveness (AHR), while ![]() enhance T cell proliferation and exacerbate AHR.182,183 Immunosuppression by MDSCs also occurs in the tumor microenvironment in which NO•-mediated increase in cGMP activation, facilitates their binding to cytotoxic lymphocytes to reduce their proliferation.184,185 Peroxynitrite and H2O2 are produced by the combined and cooperative activities of NADPH oxidase, arginase, and iNOS in different MDSC subsets. These drive several molecular blocks in T cells, ranging from the loss of TCRζ-chain expression, interference with IL-2 receptor–mediated signaling and nitration, and subsequent desensitization of the TCR.184,185 Signal transducer and activator of transcription 3 (STAT3) is a critical regulator of MDSCs. STAT3-mediated upregulation of the NADPH oxidase and ROS levels enhances the suppressive potential of MDSCs.155,184,185
enhance T cell proliferation and exacerbate AHR.182,183 Immunosuppression by MDSCs also occurs in the tumor microenvironment in which NO•-mediated increase in cGMP activation, facilitates their binding to cytotoxic lymphocytes to reduce their proliferation.184,185 Peroxynitrite and H2O2 are produced by the combined and cooperative activities of NADPH oxidase, arginase, and iNOS in different MDSC subsets. These drive several molecular blocks in T cells, ranging from the loss of TCRζ-chain expression, interference with IL-2 receptor–mediated signaling and nitration, and subsequent desensitization of the TCR.184,185 Signal transducer and activator of transcription 3 (STAT3) is a critical regulator of MDSCs. STAT3-mediated upregulation of the NADPH oxidase and ROS levels enhances the suppressive potential of MDSCs.155,184,185
Inflammasome activation is an innate immune response to pathogens, but also accompanies the development of autoimmune and chronic inflammatory diseases. Inflammasomes are multicomponent platforms that sense a variety of danger signals, including bacteria, viruses, pathogenic crystals; aggregates through a family of nod-like receptors and consists of caspase-1 to process proinflammatory cytokines for activation.186–188 Inflammasome activation starts with a priming signal followed by an activation signal. Studies with ROS scavengers suggest a role of ROS in inflammasome activation.186–190
 EPITHELIAL CELLS
EPITHELIAL CELLS
The epithelium of the airways and the alveoli is exposed to high levels of oxygen and to other environmental oxidizing species. Although inflammatory cells are major producers of RNS/ROS, resident lung cells such as epithelial cells, possess enzymatic systems for regulated production of RNS and ROS. DUOX enzymes are the primary contributors of H2O2 in the airway epithelium.25,191 DUOXs are expressed in ciliated surface cells, but not in nonciliated cells or basal cells of the upper airway. The levels of expression of DUOX1 and DUOX2 are selectively regulated by cytokines, with Th1 cytokines regulating DUOX2 and Th2 cytokines regulating DUOX1.19,21,192,193 The highly inducible DUOX2 mediates host responses to infection and inflammation, while DUOX1 is constitutively expressed in noninflamed airways; DUOX2 plays a role in innate immunity, cell signaling, and mucus production.19,21,193,194 The airway epithelium participates in innate immune response through the secretion of immune effectors such as mucin, antimicrobial peptides, and ROS to entrap or kill invading microbes. Epithelial cells use microbial pattern recognition receptors for innate immune system recognition to discriminate self from nonself.19,21,192,193 The release of cytokines/chemokines by the epithelium induces neutrophil recruitment and the activation of transcription factors augmenting the inflammatory response. Lactoperoxidase (LPO) a heme-containing peroxidase in concert with DUOX-generated H2O2 generates hypohalous acids that kill pathogens.25,194 A functional difference between the airway DUOX/LPO system and the phagocytic NOX2/MPO system is that the phagocytic system is active only during the respiratory burst, whereas DUOX generates H2O2 continuously.19,21,192,193
 ENDOTHELIAL CELLS
ENDOTHELIAL CELLS
Endothelial cells (ECs) also participate in innate immunity and crosstalk with immune cells. The importance of ECs in inflammation-induced vascular dysfunction is dependent on their ability to produce and respond to ROS and RNS. Inflammation may alter the balance between NO• and ![]() within (and surrounding) ECs, which is necessary for normal vascular function. ROS produced by the endothelium play an important role in vascular pathology.195,196 ROS can quench NO• and mediate proinflammatory signaling. Targeting ROS-quenching enzymes catalase and SOD in ECs alleviates toxic effects of excessive ROS and suppresses proinflammatory mechanisms, including endothelial cytokine activation and barrier disruption.197–199 Pulmonary EC-derived ROS play a pivotal role in EC activation and function. Alterations in EC phenotype contribute to vascular tone, permeability, and inflammatory responses and, thus, have been implicated in lung diseases, including pulmonary hypertension, ischemia-reperfusion (IR) injury, and adult respiratory distress syndrome.200–202 Contrasting effects of NOS isoforms occur during IR injury, where eNOS appears to be protective and iNOS detrimental. Under homeostatic conditions, the low NO flux generated by eNOS prevents leukocyte recruitment and associated tissue damage through scavenging of ROS. However, when iNOS expression increases, NO• levels rise and induce tissue injury.200–202 Thus, the beneficial effects of specific NOS isoforms depend on the type of primary damaging event.
within (and surrounding) ECs, which is necessary for normal vascular function. ROS produced by the endothelium play an important role in vascular pathology.195,196 ROS can quench NO• and mediate proinflammatory signaling. Targeting ROS-quenching enzymes catalase and SOD in ECs alleviates toxic effects of excessive ROS and suppresses proinflammatory mechanisms, including endothelial cytokine activation and barrier disruption.197–199 Pulmonary EC-derived ROS play a pivotal role in EC activation and function. Alterations in EC phenotype contribute to vascular tone, permeability, and inflammatory responses and, thus, have been implicated in lung diseases, including pulmonary hypertension, ischemia-reperfusion (IR) injury, and adult respiratory distress syndrome.200–202 Contrasting effects of NOS isoforms occur during IR injury, where eNOS appears to be protective and iNOS detrimental. Under homeostatic conditions, the low NO flux generated by eNOS prevents leukocyte recruitment and associated tissue damage through scavenging of ROS. However, when iNOS expression increases, NO• levels rise and induce tissue injury.200–202 Thus, the beneficial effects of specific NOS isoforms depend on the type of primary damaging event.
 FIBROBLASTS
FIBROBLASTS
Fibroblasts and fibroblast-like mesenchymal cells participate in innate immunity and in tissue repair. Such cells are typically resident within the adult human lung203; however, studies have reported fibroblasts derived from bone marrow cells204 or epithelial cells, the latter in a process known as epithelial-to-mesenchymal transition (EMT).205 EMT is a process regulating cell plasticity, which allows epithelial cells to lose their polarity and specialized junctional structures, to undergo cytoskeletal reorganization, and to acquire morphological and functional features of mesenchymal-like cells. Myofibroblasts are the primary “effector” cells in tissue remodeling and pulmonary fibrosis.206–208 Activation of the NADPH oxidase isoform, NOX4, mediates generation of H2O2, myofibroblast differentiation, contractility, and ECM production in response to TGF-β1.209 In addition, NOX4 may play a profibrotic role by inducing apoptosis of lung epithelial cells, while myofibroblasts themselves acquire an apoptosis-resistant phenotype. Epithelial cell death may also be mediated indirectly by the paracrine secretion of H2O2 by activated myofibroblasts, supporting the concept that NOX4 may be responsible for both myofibroblast activation and epithelial cell disrepair.210
OXIDATIVE STRESS IN VARIOUS LUNG DISORDERS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


