Chapter 122
Raynaud’s Phenomenon
Ariane L. Herrick, Lindsay Muir
Based on a chapter in the seventh edition by Gregory J. Landry
It is now more than 150 years since Auguste Gabriel Maurice Raynaud (1834-1881) described the phenomenon that bears his name, in his doctorate thesis of 1862: “De l’asphyxie locale et de la gangrène symétrique des extrémités”.1 Raynaud’s phenomenon (RP) is an exaggeration of the normal physiologic response of the extremities to cold exposure or emotional stress. Over the years there has been some confusion in terminology. Currently the preferred terms are as follows:
Primary Raynaud’s phenomenon (PRP): the common, idiopathic form of RP (previously sometimes termed Raynaud’s disease).
Secondary Raynaud’s phenomenon: RP secondary to an underlying cause (previously sometimes termed Raynaud’s syndrome).
Vascular surgeons may encounter patients with RP for two main reasons:
Diagnosis. The referring doctor may be concerned that the patient presenting with RP has/may have an underlying cause amenable to surgery (e.g., a cervical rib, atherosclerosis). Therefore the vascular surgeon needs to have a good understanding of the differential diagnosis of RP.
Surgical treatment of the patient with secondary RP who has progressed to digital ulceration and/or critical ischemia. Usually the patient has an underlying systemic sclerosis spectrum disorder that is unresponsive to medical (conservative) treatments. Such a patient may benefit from surgical débridement, digital (palmar) sympathectomy, and/or a revascularization procedure. A small proportion of patients require digital amputation.
This chapter discusses the pathogenesis, diagnosis, medical and surgical management, and possible future treatment strategies for RP.
Definition and Differential Diagnosis
RP is a symptom complex. The typical triphasic color change of the fingers is white (ischemia) then blue (cyanosis) and then red (reperfusion). The majority of patients with RP have “primary” (idiopathic) RP (PRP). The feet may also be affected, although patients are more likely to describe coldness than color changes, possibly because the feet are less visible to patients than their hands. Patients with PRP, by definition,2 do not progress to have irreversible tissue injury. For this reason PRP is considered a benign phenomenon and is generally thought to be due to reversible vasospasm, and not to be associated with structural vascular change.
In contrast, RP secondary to underlying disease (“secondary” RP) can progress to digital ulceration, scarring, or gangrene. Many different conditions can result in secondary RP. Box 122-1 lists the most common. This chapter focuses on PRP and systemic sclerosis–related RP because (1) PRP is by far the most common cause of RP and (2) patients with systemic sclerosis spectrum disorders are those most likely to require surgery. In patients with systemic sclerosis (also termed “scleroderma”), RP/digital vasculopathy can be extremely severe, resulting in major morbidity, pain, and disability, reflecting how systemic sclerosis is characterized by structural as well as functional abnormalities of the digital vasculature.3
Although digital ulceration and gangrene can occur in patients in whom RP is secondary to diseases other than systemic sclerosis spectrum disorders (see Box 122-1), these cases have been less well researched, and the broad principles of management are the same. However, the wider differential diagnosis must always be considered, in particular (to surgeons), the possibility of atherosclerosis. Atherosclerosis can occasionally be solely responsible for RP (consider this possibility especially if RP is asymmetrical between limbs) or can occur concomitantly with systemic sclerosis, with potentially disastrous results in terms of finger (or toe) perfusion.
Epidemiology
Studies of the prevalence of PRP have given very different results. The reason is most likely the differences in definition of PRP and also geographic location because climate influences prevalence.4 Women are more commonly affected than men, and usually present in their teens or twenties. A UK general practice–based study suggested that the prevalence of RP may be as high as 21% in women and 16% in men,5 whereas a U.S. community-based study reported prevalences of 11% in women and 8% in men.6 There are fewer data on incidence because it is more difficult to study than prevalence. A French study reported an annual incidence of 0.25%,7 comparable to results of the Framingham offspring cohort study reported by Suter et al6 (incidence was 2.2% in women and 1.5% in men over the 7-year study period).
Although secondary RP is much less common than PRP, RP has a high prevalence in patients with connective tissue diseases (see Box 122-1). This is especially true in patients with systemic sclerosis, in whom absence of RP is very unusual.8 There has been considerable discussion as to whether RP is associated with rheumatoid arthritis. A recent meta-analysis suggested a prevalence of 12.3% in patients with rheumatoid arthritis.9
The risk of digital ulceration when RP is secondary to systemic sclerosis or connective tissue disease may be as high as 50%.10
Pathogenesis
The pathogenesis of RP remains elusive. It is likely that multiple mechanisms contribute (Fig. 122-1), and that their presence and contribution vary depending on whether RP is primary or secondary and, if secondary, then to what underlying cause. Nonetheless there are some unifying principles. As a broad generalization RP results when the balance between vasodilation and vasoconstriction is disturbed in favor of vasoconstriction. However, it is worth highlighting that even in healthy control subjects, mechanisms for controlling skin temperature and blood flow11,12 are extremely complicated and incompletely understood. Therefore the pathogenesis of RP is undoubtedly complex: almost certainly both the central mechanisms originally proposed by Raynaud1 and the “local” mechanism(s) later proposed by Lewis13 are implicated. In this chapter we consider pathogenesis under the four headings “vascular,” “neural,” “intravascular,” and “other.” However, it must be recognized that these categories all interrelate.
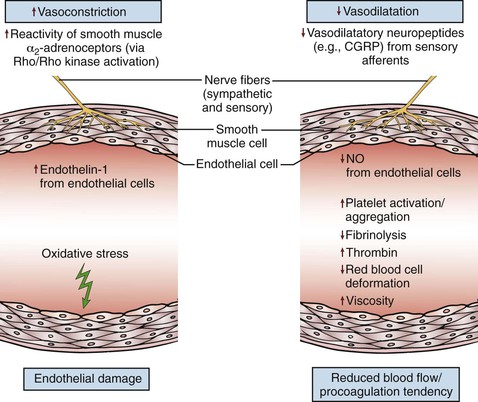
Figure 122-1 Schematic representation of some of the key elements and mechanisms contributing to the pathogenesis of Raynaud’s phenomenon. CGRP, Calcitonin gene–related peptide; NO, nitric oxide; ↑, increased; ↓, decreased. (From Herrick AL: The pathogenesis, diagnosis and treatment of primary and secondary Raynaud’s phenomenon. Nat Rev Rheumatol 8:469-479, 2012.)
An exciting point is that new insights into pathophysiology (for example, relating to nitric oxide [NO] and enthothelin-1[ET-1]) are driving new therapeutic approaches.14,15 Many of these new insights come from research into systemic sclerosis.
Vascular
Functional Abnormalities
Endothelial dysfunction is known to be one of the initial events in systemic sclerosis,16 and much of the research into vascular function in RP has focused on the endothelium. It is likely that this early endothelial involvement upsets the normal balance between vasodilation and vasoconstriction, with underproduction or reduced efficacy of vasodilators and/or overproduction of vasoconstrictors. Investigators have also studied endothelial function in PRP, comparing patients who have PRP with those who have systemic sclerosis in order to understand why those with systemic sclerosis, but not with PRP, go on to have ischemic injury. However, endothelial function has been little studied in the other conditions associated with RP.
Impaired Vasodilatation.
Endothelium-dependent vasodilatation is impaired in systemic sclerosis,14,17 and in some studies endothelium-independent vasodilatation has also been shown to be reduced.18 Although some investigators have shown reduced endothelium-dependent vasodilatation in patients with PRP,14,19,20 the evidence is weaker than for systemic sclerosis. It is not known whether production of vasodilators produced by the endothelium—including NO and prostacyclin—is reduced or whether their actions might be impaired. The situation is likely to be highly complex.21 To take NO (which relaxes smooth muscle cells) as an example, in sclerodermatous skin, constitutive endothelial NO synthase (NOS) is initially increased, but later on, in more advanced disease, inducible NOS is increased,22 and it has been suggested that NO production via neuronal NOS may be reduced in systemic sclerosis.23 Also, patients with systemic sclerosis have been shown to have increased plasma levels of an endogenous inhibitor of endothelial NOS, asymmetrical dimethylarginine.24 It is therefore unclear whether NO is underproduced or overproduced in the digits of patients with systemic sclerosis (and whether any abnormalities depend on the stage of disease). However, irrespective of this issue, NO supplementation (for example, with topical glyceryltrinitrate [GTN]) results in vasodilatation in patients with systemic sclerosis and in patients with PRP as well as in healthy controls.25
Increased Vasoconstriction.
Overproduction of vasoconstrictors by endothelial cells (e.g., ET-1 and, angiotensin II) most likely contributes to systemic sclerosis–related RP. The role of ET-1 in the pathogenesis of systemic sclerosis has received considerable attention. ET-1 is over-expressed in sclerodermatous skin.26 The evidence of a role for ET-1 in the pathophysiology of PRP is much weaker than that in systemic sclerosis. The renin-angiotensin system has been relatively little studied in patients with RP, but in systemic sclerosis, an imbalance in favor of angiotensin II is thought to occur.27
Structural Abnormalities
Structural vascular abnormalities are a well-recognized part of the systemic sclerosis disease process, affecting both the microvasculature and the digital arteries. It has been suggested that systemic sclerosis is primarily a vascular disease.28,29 These structural abnormalities, on which are superimposed the functional changes previously described, undoubtedly contribute to the impairment of digital perfusion. The microvascular abnormalities of systemic sclerosis can be clearly demonstrated noninvasively by nailfold capillaroscopy (Fig. 122-2). At the nailfold, capillaries normally run parallel rather than perpendicular to the skin surface. Typical changes are enlarged, widened capillaries, areas of avascularity, and hemorrhages. At the level of the digital artery, the most striking change is intimal hyperplasia. For reasons that are not understood, the ulnar artery is often affected in systemic sclerosis,30 with a 2012 paper suggesting that ulnar artery disease is a risk factor for digital ulceration.31Although it has been suggested that the prevalence of proximal large vessel disease is higher in patients with systemic sclerosis than in a control population,32 this issue remains controversial.
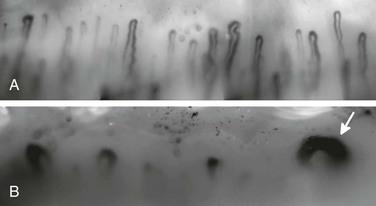
Figure 122-2 A, Normal nailfold capillaries in a healthy control subject. B, Abnormal appearances in a patient with systemic sclerosis, showing dilated (“giant”) capillaries (arrow) and large areas of a vascularity.
The pathogenesis of the structural vascular abnormalities of systemic sclerosis is not fully understood.21,33–35 Contributors are likely to include endothelial injury, endothelial cell apoptosis, abnormal expression of transcription factors, abnormal production of growth factors and cytokines, pericyte activation,36 and abnormalities both of angiogenesis—including overexpression of the antiangiogenic vascular endothelial cell growth factor VEGF165b37—and of vasculogenesis.38
As mentioned previously, structural vascular abnormalities do not occur in PRP. However, the distinction between PRP and systemic sclerosis is not absolute. One percent to 2% per year of patients who present with what initially appears to be PRP progress to a systemic sclerosis spectrum disorder or other underlying disease.39 Why the thumb is less affected by RP than other digits is not known40 but may relate to the simple observation that its shorter length might in some way be protective for structural and/or functional change.
As vascular surgeons are very aware, structural problems may occur proximally in patients with atherosclerosis and other processes such as thromboangiitis obliterans (see Chapters 5 and 79).
Neural
Several different neurotransmitters (released from either autonomic or small nerve fibers) and their receptors may be implicated in the pathogenesis of RP. The reason is that many mediate either vasodilatation or vasoconstriction.
Impaired Vasodilatation
Calcitonin gene–related peptide (CGRP) immunoreactivity is reduced in the finger skin of patients with systemic sclerosis and of those with PRP, but more so in the former.41 Other vasodilators, including substance P, neurokinin A, and vasointestinal peptide, could also be implicated but have been less studied.
Increased Vasoconstriction
The suggestion that adrenergic function is abnormal in patients with RP has attracted considerable interest over the years. The function of α2-adrenoceptors contributes more than the function of α1-adrenoceptors in the control of digital vascular tone,42 and α2-adrenoceptor responsiveness is increased in arterioles from patients with systemic sclerosis43 (and is augmented by cold).44 The α2c-adrenoceptor, which relocates from the Golgi compartment to the cell surface on cooling,45 is thought to be responsible for cold sensitivity. This relocation of the α2c-adrenoceptor is induced by Rho kinase,46 which in turn may be stimulated by reactive oxygen species produced in response to cooling.47,48 Increased protein tyrosine kinase activity and tyrosine phosphorylation may also contribute to arteriolar vasoconstriction in both PRP49 and systemic sclerosis–related RP.50
Central Mechanisms
Central mechanisms have received very little attention, despite the fact many patients describe RP in response to stress. Edwards et al51 showed that patients with PRP do not habituate in the same way as healthy controls to the alerting response (which included vasoconstriction in the cutaneous circulation of the digits) evoked by acute emotional stress.
Intravascular Factors
In some patients, intravascular factors are the cause of RP, for example, in patients with hyperviscosity syndromes (see Box 122-1). In patients with systemic sclerosis, intravascular factors are contributory rather than causal. Platelet activation, defective fibrinolysis, increased thrombin generation, reduced red blood cell deformability, white blood cell activation, and increased viscosity have all been implicated.14,52–55 Platelet activation may also occur in PRP.56,57 Activated platelets produce a number of factors, including vascular endothelial cell growth factor (which has effects on endothelial cells),58 serotonin (vasoconstrictive and profibrotic),59 and transforming growth factor (TGF)-beta and platelet-derived growth factor (profibrotic).54 Therefore platelet activation exemplifies how intravascular and vascular abnormalities interrelate and may lead to fibrosis.
Studies over the past 20 years by a number of investigators, using different methodologies, have lent support to the hypothesis that oxidative stress (which may damage endothelial cells through peroxidation of cell membranes) occurs in patients with systemic sclerosis.60–62 Oxidative stress may also occur in patients with PRP, although the evidence is weaker.
Other Mechanisms/Factors
Genetic63–65 and hormonal factors most likely also contribute to pathogenesis. Variations in blood flow during the menstrual cycle have also been described.66 The effect of estrogen is complex;21 both vasodilatory and vasoconstrictive roles have been suggested, the latter possibly via increased α2C-adrenoceptor responsiveness.67
Diagnosis
The diagnosis of RP is based on the history of color change of the fingers and therefore relies on the taking of a careful history from the patient. The more challenging question for the clinician is “Why does this patient have RP?” If there is an underlying cause (see Box 122-1), then it must be identified and treated. Conversely, if there is no underlying cause, and the RP is primary, the patient may be reassured.
The history, physical examination, and investigation of RP are in large part guided by the proposed (and generally accepted) criteria for PRP2: episodic attacks of acral pallor or cyanosis; strong and symmetrical peripheral pulses; no evidence of digital pitting, ulceration, or gangrene; normal nailfold capillaries; negative antinuclear antibody (ANA) test result; and normal erythrocyte sedimentation rate (ESR). If these criteria are not fulfilled, then the patient requires more detailed evaluation.
History
Key questions to ask during history taking are as follows:
4. Is the patient taking any drugs that could be causing RP (e.g., beta-blockers)?
6. Is there a family history (there are genetic components to both PRP and systemic sclerosis)?
Physical Examination
Key questions to answer with the physical examination are as follows:
2. Are there any features of ischemia/irreversible tissue injury (e.g., pitting scars, digital ulcers [Fig. 122-3], or evidence of previous amputations, either surgical or autoamputation)?
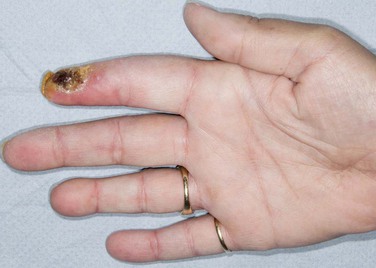
Although many of the “stigmata” of connective tissue disease described in question 3 occur in the finger and toes, a full examination must be performed because signs pointing to a connective tissue disease may be evident elsewhere (e.g., basal crackles in a patient with RP may indicate pulmonary fibrosis, which commonly occurs in connective tissue diseases).
Investigations
If the history and physical examination suggest that RP is most likely to be primary,2 then a simple set of investigations comprising a blood count, ESR, ANA, and nailfold capillaroscopy is sufficient. However, many clinicians also request a blood biochemical profile with thyroid function tests and a thoracic outlet radiograph to look for a cervical rib.
If an underlying systemic sclerosis spectrum disorder is suspected, a more comprehensive autoantibody screen should be performed, including anticentromere and antitopoisomerase (anti-Scl-70) antibodies.68 Other investigations should be requested as clinically indicated from the history and examination.
Nailfold Capillaroscopy
Ideally all patients with RP should undergo nailfold capillaroscopy. The reason is that in the patient who presents with “isolated” RP (i.e., no definite symptoms or signs of an underlying cause), nailfold capillaroscopic abnormalities as well as systemic sclerosis-specific autoantibodies have been shown to be independent risk factors for systemic sclerosis.69 Koenig et al69 monitored 586 patients with RP for 3197 patient-years. Systemic sclerosis developed in 1.8% of those with neither abnormal capillaroscopy nor a systemic sclerosis-specific autoantibody, in 25.8% with abnormal capillaroscopy, in 35.4% with a specific autoantibody, and in 79.5% with both predictors. Patients with both predictors were 60 times more likely to develop systemic sclerosis than those with neither. Partly because of these findings and those of other investigators,70,71 and partly because of improvements in methodologies, nailfold capillaroscopy is now being increasingly applied in both clinical practice and research. The aim is to identify patients with an underlying systemic sclerosis spectrum disorder at a very early stage of the disease.72,73 The presence of “scleroderma pattern” capillary abnormality, first described by Maricq et al74 and characterized by dilated loops and areas of avascularity (see Fig. 122-2), is highly specific for an underlying systemic sclerosis spectrum disorder.
Although the increased application and availability of high-magnification video capillarosopy (200-600×) has been one of the factors leading to greater interest in capillaroscopy,75,76 not all clinicians have access to it or to standard widefield microscopy. However, the more obvious nailfold capillary abnormalities may also be visualized with a dermatoscope (magnification in the order of 10×)77,78 or ophthalmoscope,79,80 although these two instruments are less likely to detect subtle early changes.
It has been suggested that in patients with systemic sclerosis, the degree of capillaroscopic abnormality is a predictor of digital ulceration.81,82 If this suggestion is confirmed, then clinical application of capillaroscopy is likely to increase.
Other Vascular Investigations
Investigation of (Proximal) Large Vessel Disease.
Patients referred to a vascular surgeon with RP may have been referred because of concerns about the possibility of proximal large vessel disease—for example, RP is unilateral (normally RP is symmetrical between limbs) or the peripheral pulses are absent or difficult to feel. In such patients, the usual first step consists of obtaining noninvasive physiologic studies and arterial duplex ultrasonography. If findings of these studies suggest large vessel disease, large vessel imaging is most likely required, with conventional (digital subtraction), magnetic resonance, or computed tomography angiography.
Thermography.
Some specialist centers include thermography (which measures surface temperature) in the investigation of patients with RP, as it can help differentiate between patients with PRP and those with systemic sclerosis–related RP (Fig. 122-4).83–85 Usually a cold challenge test is part of the thermography protocol. However, thermography is a relatively expensive technique available only in specialist centers.
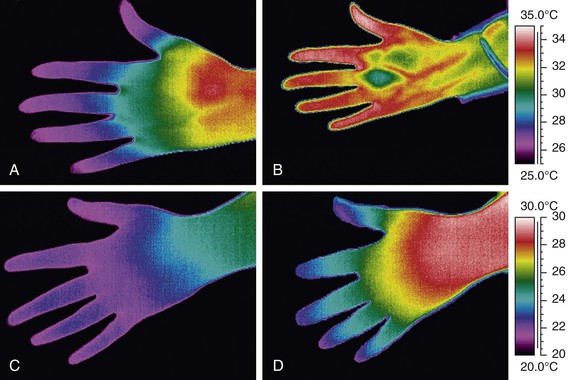
Figure 122-4 Thermograms from a patient with primary Raynaud’s phenomenon (PRP) (upper panels) and from a patient with systemic sclerosis (SSc) (lower panels). The thermograms on the left (A and C) are at 23°C, and those on the right (B and D) at 30°C. Although in both patients the fingertips are cold at 23°C, this temperature gradient along the fingers normalizes at 30°C in the patient with PRP but not in the patient with SSc (suggesting underlying structural vascular disease).
Other Laboratory Methods.
Methods other than nailfold capillaroscopy and thermography have been used to differentiate between primary and systemic sclerosis–related RP. However, these are primarily research tools, and none is widely used in clinical practice. These methods include laser Doppler flowmetry, laser Doppler imaging,86,87 plethysmography, and finger systolic pressure measurements.88 There is current interest in trying to validate some of these noninvasive methods as outcome measures for use in clinical trials of RP.
Medical Management
The main aim of treatment in patients with RP is to improve blood flow to the digits, thereby reducing the pain and discomfort of RP attacks and (in patients with secondary RP) reducing the chance of progression to irreversible digital ischemia. This aim is achieved by removing any causal or triggering factor. Doing so usually involves adjustment to environmental stimuli. Rarely, a causal factor may have a surgical solution, for example, removal of a cervical rib. It is also achieved by enhancing vasodilatation and/or reducing vasoconstriction (the rationale behind most currently used drug treatments).
Increased understanding of the underlying pathophysiological mechanisms of RP is opening up new avenues of therapy. A major advance in the past decade has been the increased international networking of clinicians with an interest in RP, with the result that large multicenter clinical trials of RP/digital ulceration are now feasible, as evidenced by several publications.89–91 Therefore the infrastructure is now in place to facilitate development of promising new therapies. This is a welcome development because there have formerly been relatively few adequately powered studies examining treatment of RP, with the result that the existing evidence upon which to base recommendations is weak.92
Treatment of RP differs among patients and depends on severity. PRP often responds to general (nondrug) measures alone and may even improve spontaneously). In contrast, the patient with systemic sclerosis and severe digital ulceration and/or critical ischemia may require hospitalization for intravenous (IV) prostanoid therapy and possibly surgery. Medical management is divided into three categories: general (nondrug) measures, drug therapy, and medical management of digital ulceration/critical ischemia. Surgical treatment, required for patients with severe digital ulceration and/or critical ischemia that does not respond to medical management, is discussed in the next section, as are botulinum toxin injections, which have now been advocated for severe digital ischemia/ulceration. The treatment approach to RP, both “uncomplicated” (no ulceration or critical ischemia) and “complicated” (progressing to ulceration or critical ischemia) is summarized in Figure 122-5.
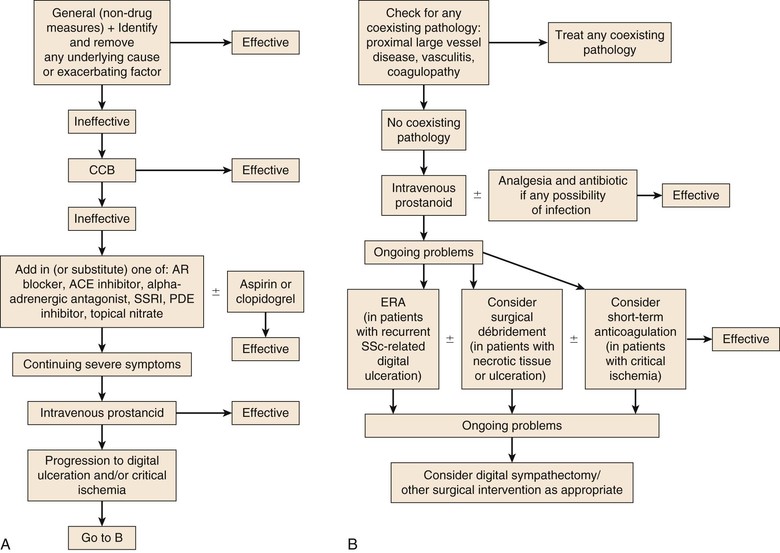
Figure 122-5 Flow charts of the approach to treatment of uncomplicated Raynaud’s phenomenon (A) and of Raynaud’s phenomenon that has progressed to digital ulceration and/or critical ischemia (B). ACE, Angiotensin-converting enzyme; AR, angiotensin receptor; CCB, calcium channel blocker; ERA, endothelin-1 receptor antagonist; PDE, phosphodiesterase; SSc, systemic sclerosis; SSRI, selective serotonin reuptake inhibitor. (From Herrick AL: The pathogenesis, diagnosis and treatment of primary and secondary Raynaud’s phenomenon. Nat Rev Rheumatol 8:469-479, 2012.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


