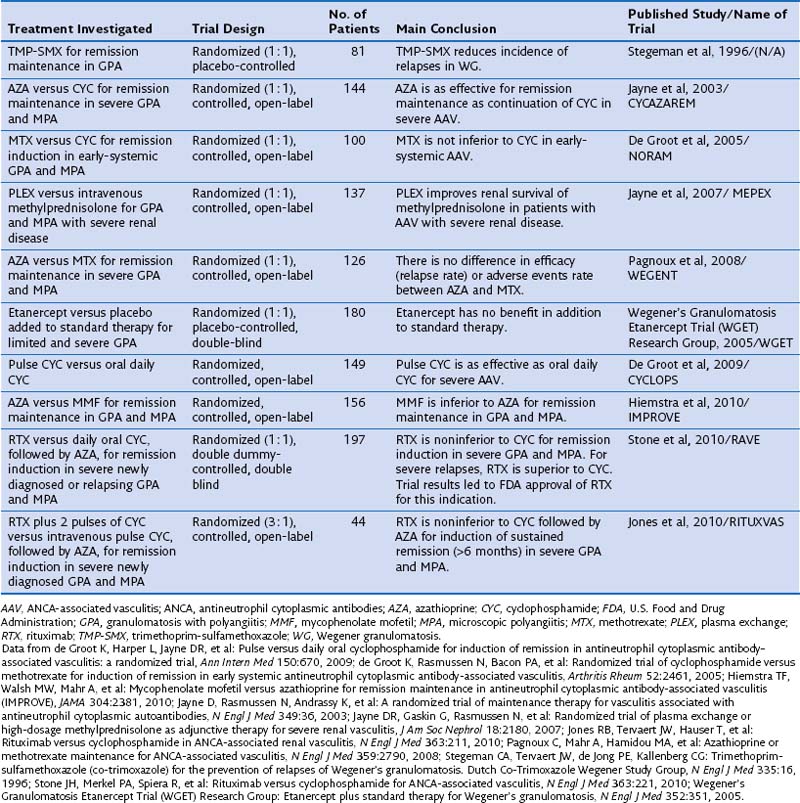
Standard therapy for GPA and MPA currently follows the same basic principles. Methotrexate (MTX) at a dose of up to 25 mg once a week in combination with oral prednisone is considered the standard of care for patients with limited GPA. However, only one prospective randomized trial has compared MTX and cyclophosphamide (CYC) for remission induction in such patients (Table 59-1). The trial, conducted by the European Vasculitis Study Group (EUVAS), showed that MTX is noninferior to CYC for remission induction, but the side effects were less frequent and less severe. The trial also documented that early discontinuation of immunosuppression in patients with ANCA-associated vasculitis is fraught with a high relapse rate. The largest reported group of patients with limited Wegener’s granulomatosis (WG) was treated with MTX for remission induction in the context of the Wegener’s Granulomatosis Trial (WGET). More than 90% of patients achieved remission with this regimen, and more than 70% achieved a sustained remission (lasting longer than 6 months). These rates are equivalent to those achieved with CYC in severe disease, as discussed next.
CYC at a dose of 2 mg/kg/day in combination with prednisone has been the standard of care for patients with severe GPA or MPA until recently. In contrast with the original regimen introduced by Fauci 40 years ago, the current consensus is to limit the duration of CYC therapy to the first 3 to 6 months of remission induction.
Once remission has been induced and the prednisone taper is well under way, CYC should be switched to either azathioprine (AZA), preferred in patients with renal involvement and any degree of renal insufficiency, or MTX. The first option is supported by the results of a randomized trial showing that AZA is as good as CYC for remission maintenance to 18 months. Another randomized controlled trial has shown that MTX and AZA are equivalent for remission maintenance. By contrast, a recent randomized controlled trial that compared mycophenolate mofetil (MMF) with AZA for remission maintenance has shown that MMF is inferior to AZA for this purpose. Thus, the use of MMF for remission maintenance can be supported only for patients who have failed MTX and AZA, or who have contraindications to both agents. The WGET study, in which MTX was used for remission maintenance, confirmed that long-term remission remains an elusive goal for many patients, because remission was maintained in less than half of the patients.
Whenever CYC is used for remission induction, consideration should be given to the patient’s fertility. Young men should be offered sperm banking before therapy is initiated. If time allows, ovarian protection should be offered to young women, in addition to minimizing the cumulative exposure as much as possible.
One randomized controlled trial has evaluated a regimen of intravenous pulse application of CYC compared with daily oral use. The pulse regimen was noninferior to the oral application of CYC for remission, and the frequency of leukopenia, but not infection, was lower. Even though the trial was not powered to detect a difference in relapses, however, the relapse rate was higher after remission induction with the intermittent pulse regimen than with the oral application of CYC.
A metaanalysis of earlier cohort studies had also indicated that intravenous pulse CYC therapy may be safer because of a lower cumulative dose, but a higher relapse rate also was observed after discontinuation. In my own experience, intravenous CYC is best avoided in the intensive care unit setting. However, its use is preferred over oral CYC in patients with questionable compliance, in young women with fertility issues, and in patients who have gastrointestinal problems with oral CYC application.
The four-decade-old standard use of CYC for remission induction in patients with severe GPA and MPA recently has been challenged by the results of two randomized controlled trials. The RAVE (rituximab for ANCA-associated vasculitis) trial was a randomized double-blind, double-placebo-controlled, multicenter trial that compared oral CYC (2 mg/kg/day) to rituximab (RTX) (375 mg/m2 body surface area/week for 4 weeks) for remission induction in severe GPA or MPA in 197 patients. Once remission was achieved between 3 and 6 months, patients randomized to receive CYC were switched to AZA for remission maintenance for 18 months, whereas patients in the original RTX group then received placebo. No difference was observed in rates of achieving remission at the end of 6 months and maintaining remission at 18 months between the two treatment arms. Among the 101 patients who entered the trial with severe relapsing disease, however, RTX proved superior to CYC. The results of the RAVE trial led to approval by the U.S. Food and Drug Administration (FDA) of RTX for remission induction in severe GPA and MPA.
Another randomized controlled open-label trial conducted in 44 patients with newly diagnosed severe ANCA-associated vasculitis with active renal disease, RITUXVAS (Rituximab versus Cyclophosphamide for ANCA-Associated Renal Vasculitis), showed results complementary to those of the RAVE trial. In the RITUXVAS trial, patients were randomized 3 : 1 to receive RTX (together with two pulses of CYC) compared with standard intravenous pulse CYC therapy followed by oral AZA. The primary outcome was sustained remission (of more than 6 months’ duration) at month 12. No difference between the treatment groups was found (RTX 76% versus CYC 82%).
Mycophenolate mofetil (MMF) may represent an alternative to CYC (and RTX) for patients with MPA who have MPO-ANCA and mild renal disease (as defined by creatinine levels less than 3.5 mg/day) and no other life- or organ-threatening disease manifestation. This claim is supported by data from a randomized controlled trial in 35 patients from China comparing oral MMF (1.5 to 2 g/day) with intravenous CYC (0.75 to 1.0 g/m2 once monthly). In addition, all patients received intravenous methylprednisolone bolus therapy (0.5 g/day for 3 days) followed by oral prednisone (0.6 to 0.8 mg/kg for 4 weeks tapered by 5 mg every week to reach 10 mg/day). These regimens demonstrated equivalent efficacy, but MMF was better tolerated than CYC. A prospective pilot trial in 17 patients conducted at the Mayo Clinic achieved similar results.
Only gold members can continue reading.
Log In or
Register to continue
Related