Chapter 47 Idiopathic Pulmonary Fibrosis and Other Interstitial Lung Diseases
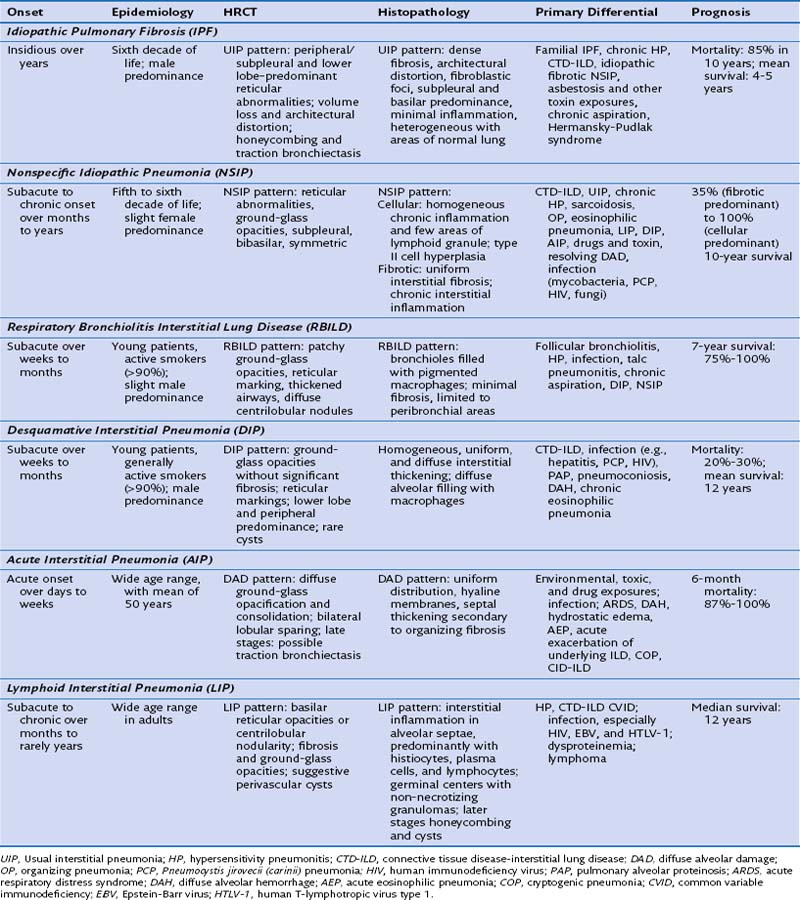
These ILDs differ based on epidemiology, onset of disease, chest imaging findings, histopathology, and most importantly, prognosis and response to treatment. Cryptogenic organizing pneumonia presents and is treated similarly to noncryptogenic presentations of organizing pneumonia, as discussed in Chapter 50. The remaining idiopathic ILDs are discussed in this chapter. Table 47-1 compares various characteristics and Figure 47-1 shows some distinct radiologic and histologic patterns of these idiopathic ILDs.
Idiopathic Pulmonary Fibrosis
1. Extensive fibrosis and architectural distortion, characterized by significant myofibroblasts and fibroblasts and dense collagen in the interstitium. This may be with or without honeycombing.
2. Heterogeneous involvement of the lung with areas of diseased lung, mostly in the subpleural/basilar regions, intermixed with areas of normal lung.
4. Lack of features inconsistent with UIP:
1. Subpleural/basal predominance
2. Honeycombing with or without traction bronchiectasis
4. Lack of features inconsistent with UIP:
1. The clinical context is that of an idiopathic ILD; other potential causes or associations, including infection, drug or environmental exposures, and systemic disorders (e.g., CTDs), have been excluded.
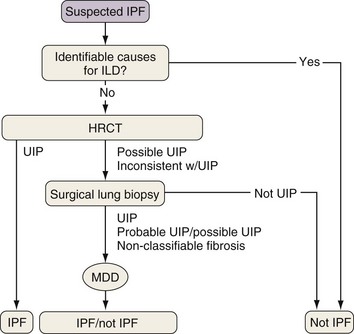
Because the diagnosis of IPF requires combining clinical, radiologic, and often pathologic information, multidisciplinary discussions with chest imagers and pathologists are strongly recommended, especially since a definitive diagnosis cannot be made based on radiologic and histologic findings alone. Figure 47-2 outlines an algorithm for the diagnosis of IPF.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


