Pulmonary Manifestations of the Collagen Vascular Diseases
Pleuropulmonary involvement associated with the collagen vascular diseases occurs frequently. All the structures within the respiratory tract may be affected, either separately or in combination. This includes the respiratory muscles, the pleura, the conducting airways, and the lung parenchyma—the small airways, the interstitium, or the pulmonary vessels. Moreover, these patients experience an increased incidence of community-acquired pneumonia as well as pneumonia associated with the immunosuppressive drugs employed for treatment. Anti–tumor necrosis factor-α (anti–TNF-α) agents increase the risk for infections, particularly mycobacterial pathogens, both tuberculous and nontuberculous. Cytotoxic drugs, particularly methotrexate and gold, can also induce various noninfectious interstitial reactions, which are often difficult to distinguish from a primary interstitial complication of a collagen vascular disease.1
Although most pulmonary complications appear in an established case of a collagen vascular disease, lung disease may precede the more typical systemic manifestations.2 For example, in both rheumatoid arthritis and polymyositis–dermatomyositis, the interstitial lung disease may precede the joint and muscle disease for several months to several years. This is also the case, but to a lesser extent, for scleroderma. In one study, 19% of patients initially diagnosed with idiopathic pulmonary fibrosis developed a collagen vascular disease over a period of 1 to 11 years, primarily rheumatoid arthritis or polymyositis–dermatomyositis. These individuals were younger and more likely to be women. Pleuritis with or without effusion sometimes heralds the onset of rheumatoid arthritis or systemic lupus erythematosus (SLE). An acute immunologic pneumonitis or diffuse alveolar hemorrhage has been reported to be the signal event in SLE, polymyositis–dermatomyositis, and mixed connective-tissue disease.
The incidence of the pleuropulmonary complications (Table 60-1) is variable. Interstitial lung disease is reported to be as high as 60% in premortem and 100% in postmortem studies in scleroderma. In contrast, interstitial lung disease in ankylosing spondylitis is an uncommon event. In general, the incidence of interstitial lung disease is increasing for most of the collagen vascular diseases, primarily due to increased recognition and more sensitive screening techniques such as high-resolution computed tomography and bronchoalveolar lavage, which will detect abnormalities in both asymptomatic as well as symptomatic patients with normal chest radiographs. Prior studies assessing the incidence of disease relied on physiologic testing, which included spirometry, lung volumes, and diffusing capacity but did not measure rest and exercise gas exchange, which is the most sensitive physiologic marker of interstitial lung disease and pulmonary vascular disease.
PULMONARY PARENCHYMAL, VASCULAR, AND AIRWAY PATHOLOGY IN THE COLLAGEN VASCULAR DISEASES
The spectrum of pathologic changes in the lung parenchyma, pulmonary vasculature, and airways are considered below, prior to a discussion of clinical findings in individual collagen vascular diseases.
 INTERSTITIAL LUNG DISEASE
INTERSTITIAL LUNG DISEASE
Interstitial involvement is a common manifestation of the collagen vascular disorders, presenting with a number of different inflammatory responses within the lung. Each response may represent a different form of lung injury or response to injury. Defining which response is underlying a patient’s interstitial lung disease has important prognostic and therapeutic significance.
Diffuse alveolar damage (DAD) is the underlying histologic lesion that is also seen in the acute respiratory distress syndrome, idiopathic acute interstitial pneumonitis (Hamman–Rich syndrome), severe viral pneumonias, and cytotoxicity from some drugs. This damage consists of a mixed interstitial inflammatory infiltrate, interstitial edema and fibrin deposition, and characteristic intra-alveolar hyaline membrane formation. Intra-alveolar red blood cells (diffuse alveolar hemorrhage) may be present in severe cases. With progression, there is intra-alveolar organization, intra-alveolar and interstitial fibrosis, alveolar collapse, and the development of an end-stage fibrotic or “honeycomb” lung. An acute immunologic pneumonia, seen in SLE (acute lupus pneumonitis) and in polymyositis–dermatomyositis, may also demonstrate this underlying histologic appearance.
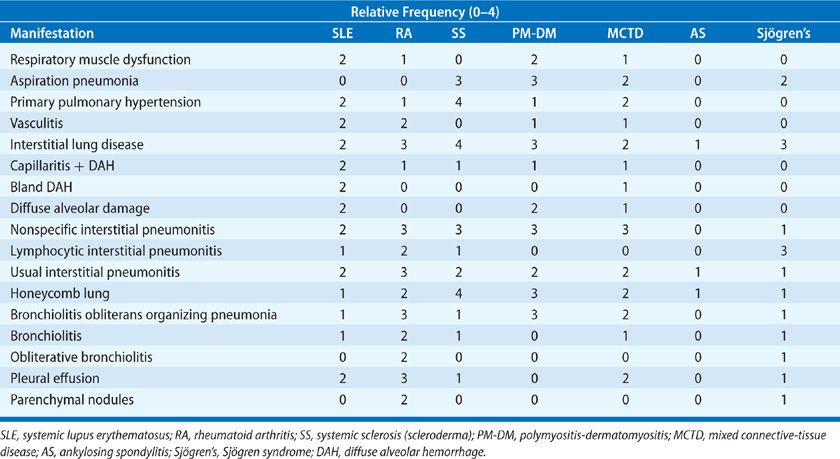
Nonspecific interstitial pneumonitis (NSIP) refers to a spectrum of histologic features with varying degrees of lymphoplasmacytic infiltration of the interstitium and collagen deposition (Fig. 60-1). In the cellular form, lymphoplasmacytic interstitial inflammation exists with associated type II alveolar epithelial cell hyperplasia. In the fibrosing form, the inflammation is accompanied by a temporally and spatially homogeneous deposition of collagen (fibrosis). Architectural distortion or honeycombing may occur in advanced cases and the presence of fibrosis dramatically changes the clinical course and prognosis to one resembling that seen in usual interstitial pneumonitis (UIP) (see below). NSIP is most frequently seen in patients with rheumatoid arthritis, polymyositis–dermatomyositis, mixed connective-tissue disease, and scleroderma.
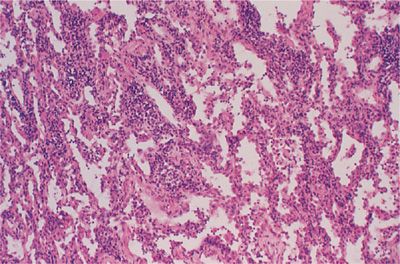
Figure 60-1 Nonspecific interstitial pneumonitis (NSIP) in rheumatoid arthritis. There is a lymphoplasmacytic infiltration of the interstitial compartment with minimal collagen deposition.
Lymphocytic interstitial pneumonitis refers to a monotonous infiltration of the interstitium by mature lymphocytes (Fig. 60-2). These lymphocytes tend to form germinal centers within the interstitium as well as displaying an angiocentric distribution. Other features of lymphocytic interstitial pneumonia include macrophagic giant cells, granuloma formation, and amyloid deposition. Lymphocytic interstitial pneumonitis can progress to UIP and end-stage honeycomb lung. Among the collagen vascular diseases, this pneumonitis most commonly accompanies the primary form of Sjögren syndrome and, to a lesser extent, the secondary form of Sjögren syndrome appearing with other collagen vascular diseases, particularly rheumatoid arthritis.
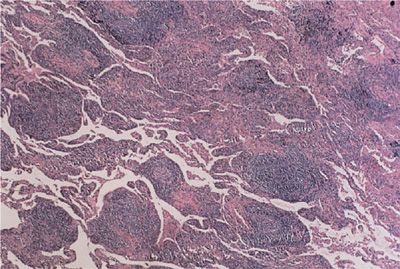
Figure 60-2 Lymphocytic interstitial pneumonitis in a patient with primary Sjögren syndrome. There is a dense lymphocytic infiltrate, broadening the interstitium and lymphoid follicles.
UIP is the underlying lesion of idiopathic pulmonary fibrosis and can also appear in all the collagen vascular diseases. It consists of varying degrees of mononuclear cell infiltration and fibroblastic proliferation leading to collagen deposition within the alveolar interstitium (Fig. 60-3). With progression, this fibrotic reaction results in marked distortion of the lung architecture and what remains are 2- to 3-mm cystic spaces lined by metaplastic epithelium, the so-called honeycomb lung (Fig. 60-4). Other features of UIP include type II epithelial cell hyperplasia producing a “hob-nailed” appearance on the alveolar surface, collections of intra-alveolar macrophages, and smooth-muscle proliferation within the interstitium. Additional abnormalities seen in collagen vascular disease-associated UIP but not in patients with idiopathic pulmonary fibrosis may include: focal chronic pleuritis, lymphoid follicles with germinal center formation, perivascular collagen deposition, and an increase in CD4+ T lymphocytes, especially in rheumatoid arthritis.
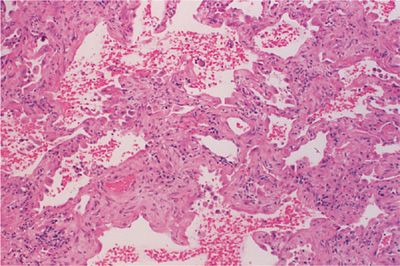
Figure 60-3 Usual interstitial pneumonia (UIP) in a patient with rheumatoid arthritis. There is broadening of the interstitium by varying degrees of mononuclear cell infiltration and collagen deposition.
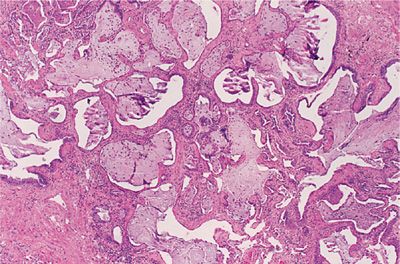
Figure 60-4 Advanced UIP in a patient with scleroderma (honeycomb lung). Normal alveolar tissue is replaced with broad bands of fibrous tissue lined by metaplastic epithelium and filled with inspissated mucus producing a cyst-like network. (Used with permission of the Armed Forces Institute of Pathology [AFIP].)
Organizing pneumonia, previously termed bronchiolitis obliterans organizing pneumonia, is a distinctive histologic lesion that follows a variety of insults to the alveolar structures including drugs, infection, radiation, and an idiopathic variety. Organizing pneumonia can also complicate the collagen vascular diseases, particularly rheumatoid arthritis and polymyositis–dermatomyositis. Three features comprise the histologic picture: (1) intra-alveolar space and intra-alveolar ductal fibroblastic proliferation with early collagen deposition (Masson bodies), (2) inflammatory polyps consisting of fibroblasts and mononuclear cells protruding into the lumens of respiratory and terminal bronchioles, and (3) alveolar septal lymphoplasmacytic infiltrate with type II pneumocyte hyperplasia within affected areas (Fig. 60-5). Bronchiolitis obliterans organizing pneumonia has the potential for being a completely reversible lesion; however, with continuing injury it may progress to end-stage fibrosis and honeycomb lung.
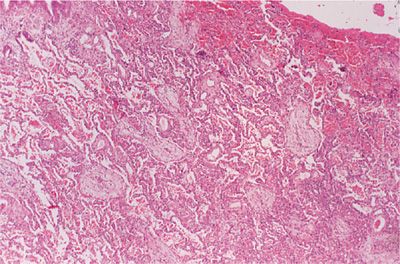
Figure 60-5 Bronchiolitis obliterans organizing pneumonia in a patient with rheumatoid arthritis. There is a mononuclear cellular infiltration of the interstitium without collagen deposition as well as alveolar duct and intra-alveolar fibroblastic proliferation and early collagen production.
 PULMONARY VASCULAR DISEASE
PULMONARY VASCULAR DISEASE
A form of pulmonary artery hypertension, which most commonly appears in patients with scleroderma and is now being increasingly recognized in SLE, rheumatoid arthritis, and mixed connective-tissue disease, is histologically identical to the syndrome of idiopathic pulmonary artery hypertension (IPAH) seen in young women without collagen vascular disease, formerly known as primary pulmonary hypertension. This is a proliferative disorder (plexogenic arteriopathy) affecting the arterioles and small muscular pulmonary arteries. This form of pulmonary hypertension must be differentiated from secondary forms as a result of hypoxic vasoconstriction induced by interstitial lung disease or severe emphysema. In the plexogenic variety, there is endothelial cell intimal proliferation and smooth muscle cell proliferation causing medial thickening with a resultant “onion ring” configuration and luminal obliteration. In the secondary forms of pulmonary hypertension due to hypoxia, medial hypertrophy is the primary finding. In patients with SLE and the antiphospholipid syndrome, pulmonary artery hypertension may develop as a result of recurrent pulmonary emboli and mimic the clinical picture of IPAH.
Vasculitis refers to an acute inflammatory angiodestructive process resulting in fibrinoid necrosis of the vascular wall. In the collagen vascular diseases, this is most often a small-vessel vasculitis involving arterioles and small muscular pulmonary arteries. Although uncommon, this is seen with greatest regularity in SLE and less frequently in rheumatoid arthritis, polymyositis–dermatomyositis, and mixed connective-tissue disease. Often accompanying the arteriolitis is the lesion of pulmonary capillaritis (see below).
 DIFFUSE ALVEOLAR HEMORRHAGE
DIFFUSE ALVEOLAR HEMORRHAGE
Diffuse alveolar hemorrhage is recognized by the accumulation of red blood cells within the alveolar spaces, and with recurrent episodes, intra-alveolar and interstitial hemosiderin is deposited and fibrosis may result. There are two different histologic subtypes seen in diffuse alveolar hemorrhage. One is devoid of inflammation and is referred to as bland hemorrhage (Fig. 60-6). It is therefore similar in histologic appearance to idiopathic pulmonary hemosiderosis. The other, pulmonary capillaritis is a unique neutrophilic infiltration of the alveolar interstitium, which results in necrosis and loss of integrity of the alveolar–capillary basement membrane, capillary destruction and thrombosis, and a leakage of red blood cells into the alveolar space (Fig. 60-7). A unique feature in pulmonary capillaritis is that many of the infiltrating neutrophils are undergoing fragmentation (leukocytoclasis), and others appear as densely staining apoptotic cells. Nuclear debris (“dust”) subsequently accumulates within the necrotic, edematous interstitium, and intra-alveolar compartments while red blood cells freely leak into the interstitial matrix due to capillary destruction. Capillary and arteriolar thrombosis, organizing pneumonia, and type II epithelial cell hyperplasia may also be seen.
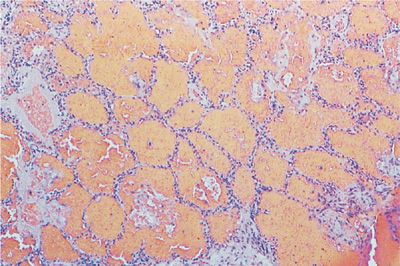
Figure 60-6 Bland diffuse alveolar hemorrhage in SLE. There is little if any interstitial reaction except for type II pneumocyte epithelial cell hyperplasia. The alveolar spaces are filled with red blood cells.
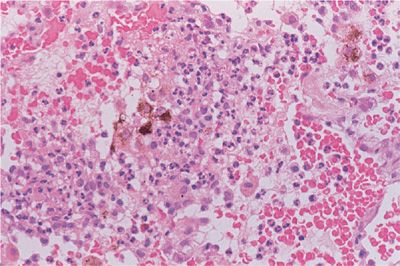
Figure 60-7 Low-power view of pulmonary capillaritis in a patient with SLE. There is marked thickening of the interstitial compartment and infiltration by acute and chronic inflammatory cells. The alveolar spaces are filled with red blood cells and neutrophils.
Capillaritis is most commonly seen in the systemic vasculitides, particularly granulomatosis with polyangiitis (GPA/Wegener granulomatosis) and microscopic polyangiitis (MPA), the small-vessel variant of polyarteritis nodosa. Of the collagen vascular diseases, both bland pulmonary hemorrhage and diffuse alveolar hemorrhage secondary to pulmonary capillaritis appear most commonly in SLE. Cases of pulmonary capillaritis have also been reported to occur in rheumatoid arthritis, Sjögren syndrome, polymyositis–dermatomyositis, and mixed connective-tissue disease.
 BRONCHIOLITIS
BRONCHIOLITIS
Bronchiolitis refers to an inflammatory-fibrotic process involving the terminal and respiratory bronchioles and possibly the surrounding alveolar structures. Respiratory bronchiolitis is primarily seen in smokers with or without an associated collagen vascular disease. There is also a primary form of cellular bronchiolitis that complicates the collagen vascular diseases, most often appearing in rheumatoid arthritis and Sjögren syndrome. Histologically, there is a mononuclear cell infiltration of the wall of the bronchiole without impingement of the bronchiolar lumen. In contrast, in bronchiolitis obliterans, or obliterative bronchiolitis, there is a concentric fibrous obliteration of the bronchiolar lumen leading to a severe obstructive lung disease (Fig. 60-8). Bronchiolitis obliterans is most often reported as a complication of rheumatoid arthritis.
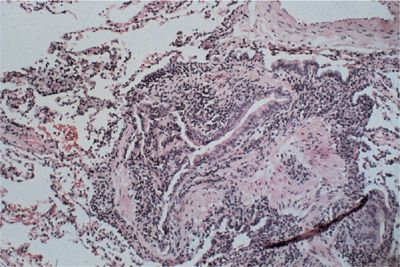
Figure 60-8 Obliterative bronchiolitis in rheumatoid arthritis. There is a marked reduction of the luminal diameter due to concentric fibrous obliteration and dense chronic inflammation. (Reproduced with permission from Schwarz MI, Lynch DA, Tuder R. Bronchiolitis obliterans: The lone manifestation of rheumatoid arthritis. Eur Respir J. 1994;7(4):817–820.)
 PARENCHYMAL NODULES
PARENCHYMAL NODULES
Noninfectious inflammatory parenchymal nodules occur in both rheumatoid arthritis and Sjögren syndrome. In rheumatoid arthritis the nodules are referred to as the necrobiotic or rheumatoid nodules. These lesions are found both in the pleura and lung parenchyma and are identical in appearance to a subcutaneous rheumatoid nodule. In the lung parenchyma, these nodules are located in the interlobular septa and in the subpleural parenchyma. The necrobiotic nodule is comprised of palisading histiocytes, giant cells, and other mononuclear cells surrounding an area of fibrinoid debris (Fig. 60-9). In Sjögren syndrome, a rounded lesion known as pseudolymphoma can occasionally be detected on the chest radiograph. Pseudolymphoma is considered to be a localized form of lymphocytic interstitial pneumonia and is made up of a dense infiltrate of lymphocytes and histiocytes with occasional granuloma formation. There is a potential risk for malignant transformation in pseudolymphoma as well as in the other forms of lymphocytic interstitial pneumonia.
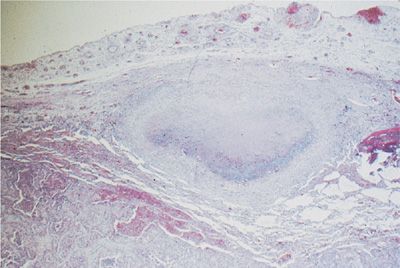
Figure 60-9 Typical subpleural location of a necrobiotic rheumatoid nodule. There is a central area of fibrinoid debris surrounded by palisading histiocytes.
SYSTEMIC LUPUS ERYTHEMATOSUS: CLINICAL FEATURES
SLE is characterized by the production of antibodies against various cellular antigens derived from the nucleus, cytoplasm, or cell membrane. Tissue injury appears to be associated with the development of immune complexes, the presence of low serum complement levels, and the production of antibodies to native DNA. Pleuropulmonary involvement in SLE occurs in the vast majority of patients (97.8%), with pleuritis (77%), bacterial infections (58%), and diffuse alveolar hemorrhage (26%) being the most common.3,4 Lung injury is thought to be the result of an immune complex-mediated injury and appears to increase in incidence depending on the age of the patient, duration of illness, pleuritis, and the presence of specific autoantibodies. Pulmonary vascular and shrinking lung syndrome appear to be associated with the development of the anti-RNP autoantibody.5 A number of syndromes (Table 60-2) are associated with acute respiratory-type illness in SLE. Patients with SLE who present with a febrile illness, cough with or without productive sputum, and new pulmonary infiltrates must be considered to have an infectious pneumonia, although acute lupus pneumonitis and diffuse alveolar hemorrhage may have a similar presentation.6 Infection can be community-acquired or a complication of immunosuppressive therapy. Infectious pneumonia represents the most common cause of pulmonary disease in SLE, and infections in general represent the most common reason for death (33%–77%).7 Bronchoalveolar lavage is often helpful in excluding an infectious pneumonia in the immunocompromised SLE patient.
Another important consideration in an acutely dyspneic SLE patient is pulmonary embolism, a complication reportedly occurring in up to 25% of patients and a significant cause of mortality.8 The occurrence of thromboembolic disease correlates with the presence in the serum of acquired antiphospholipid antibodies (lupus anticoagulant or anticardiolipin).9 The most common epitope(s) to which antibodies exist in these patients is β2-glycoprotein I. A more appropriate term may therefore be anti-β2–glycoprotein syndrome. Up to a third of patients with SLE have the antiphospholipid syndrome. Thrombocytopenia, recurrent venous or arterial thrombosis, hemolytic anemia, leg ulcers, and recurrent fetal loss are also manifestations of antiphospholipid syndrome.
Other causes for acute respiratory failure in patients with SLE include a volume overload state, due either to renal failure or to congestive heart failure secondary to myocarditis. Uremic pneumonitis with underlying DAD is also a possible cause of an acutely dyspneic SLE patient with renal failure. A syndrome, acute reversible hypoxemia, occurring in acutely ill SLE patients who are experiencing systemic exacerbations has been described.10 These patients have hypoxemia and a widened alveolar–arterial oxygen gradient, but both the chest radiograph and ventilation–perfusion lung scans are normal. It is postulated that there is complement-activated neutrophil aggregation in the pulmonary vasculature. The hypoxemia improves with immunosuppressive therapy. Given the high incidence of antiphospholipid syndrome in SLE, acute reversible hypoxemia should be considered only after excluding thromboembolic disease.
 ACUTE LUPUS PNEUMONITIS
ACUTE LUPUS PNEUMONITIS
Acute lupus pneumonitis is a clinical syndrome with an underlying histology of DAD, bronchiolitis obliterans organizing pneumonia, NSIP, or a combination of these.11 Acute lupus pneumonitis mimics an acute infectious pneumonia and may be the presenting manifestation of SLE in up to 50% of patients. In those with an established diagnosis, it also appears during a flare-up of the other systemic manifestations of SLE, particularly pleuritis, pericarditis, arthritis, and nephritis. Acute lupus pneumonitis is reportedly more common in the postpartum period. It frequently recurs and cases have been documented that have progressed to a more chronic interstitial lung disease (UIP). Fortunately, acute lupus pneumonitis is a relatively uncommon complication, occurring in less than 5% of patients.
Bilateral alveolar infiltrates, which can be patchy or densely consolidated and often accompanied by pleural effusions and cardiomegaly due to underlying pericardial effusion or myocarditis (Fig. 60-10A), are present on chest radiographs at presentation. White blood cell counts and sedimentation rates are elevated and serum complement is often low. Immunopathologic studies reveal the presence of complement as well as antibodies to IgG and DNA in some patients, supporting the concept of an immune complex pathogenesis (Fig. 60-10B). Because of the difficulty in distinguishing acute lupus pneumonitis from an infectious pneumonia, a bronchoalveolar lavage and sometimes an open (thoracoscopic) lung biopsy are indicated prior to instituting anti-inflammatory and immunosuppressive therapy. Acute respiratory failure in acute lupus pneumonitis often requires assisted mechanical ventilation. The mortality rate has been reported to be as high as 50%, with the causes of death in patients with acute lupus pneumonitis being due to respiratory failure, another complication of SLE (nephritis, cerebritis), or a superimposed infection.
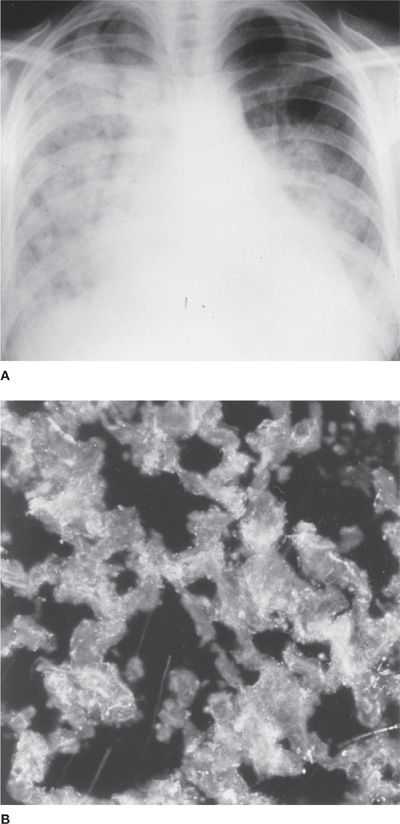
Figure 60-10 Acute lupus pneumonitis. A. The chest radiograph demonstrates diffuse alveolar filling with cardiomegaly (pericardial effusion vs. myocarditis). There is also a left pleural effusion. B. The immunofluorescent study demonstrates granular immune complex deposition in the alveolar interstitium.
 DIFFUSE ALVEOLAR HEMORRHAGE
DIFFUSE ALVEOLAR HEMORRHAGE
Diffuse alveolar hemorrhage, although rare, may be a presenting manifestation of SLE. In several cases, recurrent diffuse alveolar hemorrhage was present for years prior to the diagnosis of SLE.12 The majority of cases, in contrast to acute lupus pneumonitis, first appear in a well-documented case of SLE. Diffuse alveolar hemorrhage accounts for 1% to 4% of SLE-related hospitalizations.
Diffuse alveolar hemorrhage can also present with symptoms reminiscent of an infectious pneumonia or acute lupus pneumonitis, and the additional symptom of hemoptysis raises the possibility of this diagnosis. Hemoptysis is present in 30% to 50% of patients during their initial presentation, but up to 90% will have hemoptysis during their subsequent course. Routine laboratory work demonstrates a falling hematocrit, and in 60% to 90% of patients an active glomerulonephritis is invariably present. A progressive serosanguineous bronchoalveolar lavage may be the first clue to this diagnosis. Diffuse alveolar infiltrates are present on chest radiography (Fig. 60-11), but in contrast to acute lupus pneumonitis, pleuritis and pericarditis are not prominent features. Pathologic changes that are reminiscent of both acute lupus pneumonitis (DAD and NSIP) and diffuse alveolar hemorrhage with or without pulmonary capillaritis are not unusual in a single biopsy specimen.13 The mortality rate is approximately 50% and is independent of the underlying histopathology (bland hemorrhage vs. pulmonary capillaritis). Recurrent DAH is common and should be expected in the absence of therapeutic intervention.
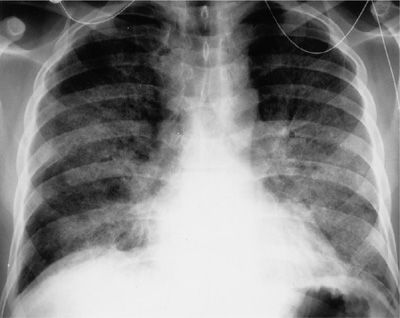
Figure 60-11 Diffuse alveolar hemorrhage in SLE. There are diffuse alveolar infiltrates without cardiomegaly or pleural effusions.
There are no controlled clinical trials for the treatment of either acute lupus pneumonitis or diffuse alveolar hemorrhage. Once infection has been excluded, corticosteroids are the mainstay of therapy. Intravenous methylprednisolone, 1 to 2 g daily in divided doses for 3 to 4 days before tapering, should be considered. Concomitant oral or parenteral cyclophosphamide or azathioprine is commonly administered, given the associated incidence of lupus nephritis. Plasmapheresis, immunoglobulin therapy, and rituximab have been used successfully, consistent with the proposed immune complex pathogenesis.14–17
 LUPUS PLEURITIS
LUPUS PLEURITIS
Pleurisy and pleural effusion are the most common primary pulmonary complications of SLE, occurring in 50% to 80% of patients. Pleurisy and/or a pleural effusion may also be the presenting and sole manifestation of the disease. They are usually recurrent and may accompany more severe complications such as acute lupus pneumonitis or nephritis. Patients complain of pleuritic pain, fever, and dyspnea. The chest radiograph may be normal (dry pleurisy) or demonstrate small to moderate pleural effusions (massive effusions are rare), which are bilateral in 50% of patients. When unilateral, there is no predilection for either side.
Effusions are serous or serosanguineous and exudative in nature. The white cell counts range from 5 to 10,000 cells/mm.3 Early on, neutrophils predominate, but with time mononuclear cells appear. These characteristics are nonspecific and are often seen with infectious parapneumonic effusions. In contrast to rheumatoid arthritis, the pleural fluid glucose concentration in lupus pleuritis is not reduced. Rheumatoid factor may be positive in lupus pleuritis. Pleural fluid complement levels are abnormally decreased in lupus, with both total complement activity and C3 and C4 reduced. A positive double-stranded pleural fluid DNA titer is nonspecific as opposed to the serum test, since it has been found in pleural effusions due to malignancy and tuberculosis. The most helpful measurement is the pleural fluid antinuclear antibody titer. Levels greater than 1:160 are very suggestive of lupus pleuritis. Examination of the pleural tissue reveals infiltration with plasma cells and lymphocytes, and, with repeated episodes, pleural fibrosis supervenes. Occasionally, a vasculitis of the pleural vessels is detected, and immune complex deposition has been reported. Corticosteroid treatment is effective for relief of pleural pain, but time to resolution of the pleural effusion is quite variable and probably unaffected by this treatment. In the unusual case, recurrent lupus pleuritis may result in massive pleural fibrosis and lung entrapment, necessitating a pleural stripping procedure.
While pleural effusions and pleurisy are common in patients with SLE, a broad differential diagnosis should be considered. The increased incidence of infectious complications, thromboembolic disease, and pulmonary hypertension in SLE predisposes patients to parapneumonic effusions and empyema, congestive heart failure, and effusions secondary to thromboembolic disease.
 INTERSTITIAL LUNG DISEASE
INTERSTITIAL LUNG DISEASE
Clinically significant interstitial lung disease is an uncommon pulmonary manifestation in SLE but UIP, lymphocytic interstitial pneumonitis, NSIP, and organizing pneumonia have all been reported. UIP is known to appear following acute lupus pneumonitis and in some cases has been documented to appear as an independent insidious disease. Using high-resolution computed tomography, 38% of patients with SLE patients with normal chest radiographs demonstrate pulmonary abnormalities consistent with some form of interstitial lung disease. In those who develop interstitial lung disease, a prior episode of acute lupus pneumonitis and an insidious onset of dyspnea are often noted. The prevalence of interstitial lung disease is increased in the subset of SLE patients with features suggestive of a mixed connective-tissue disease.
In patients who develop the insidious form of interstitial lung disease, the diagnosis of SLE is present for several years, and no other pattern of organ involvement predicts its appearance. These patients have progressive dyspnea and cough with interstitial infiltration on the chest radiograph. High-resolution computed tomography indicates combinations of ground-glass attenuation, inter- and intralobular septal thickening, and honeycomb change. Pulmonary function tests reveal a restrictive pattern with reduction in the diffusing capacity and hypoxemia accentuated by exercise. Response to therapy, either corticosteroids alone or in combination with cyclophosphamide or azathioprine, depends upon the underlying histology. Those cases with underlying NSIP or organizing pneumonia are more likely to respond to treatment than those who demonstrate excess collagen deposition and cystic honeycomb formation.
 PULMONARY VASCULAR DISEASE
PULMONARY VASCULAR DISEASE
Idiopathic pulmonary hypertension due to plexogenic arteriopathy was previously thought to be an uncommon complication of SLE. It is now estimated to occur in 1% to 9% of patients.18,19 This form of pulmonary hypertension is associated with Raynaud phenomenon, digital vasculitis, serositis, antibodies to ribonucleoprotein, rheumatoid factor, antiphospholipid antibodies, and most recently antiendothelial cell antibodies. Patients complain of dyspnea and fatigue but have normal chest radiographs. In advanced cases, pulmonary arterial enlargement appears. Spirometry and lung volumes are normal, but there is often an isolated reduction of the diffusing capacity for carbon monoxide as well as gas exchange abnormalities. Ventilation–perfusion lung scanning and, occasionally, pulmonary arteriography are indicated, particularly in those patients with the antiphospholipid syndrome who have a potential for recurrent small pulmonary emboli. Therapeutic options include vasodilator therapy, anticoagulation, immunosuppression with cyclophosphamide, and transplantation.
Vasculitis in SLE is more likely to be discovered in lung biopsy specimens, demonstrating either diffuse alveolar hemorrhage or acute lupus pneumonitis as opposed to being an isolated finding. Autopsy series indicate small-vessel vasculitis in 20% of cases.
 BRONCHIOLITIS
BRONCHIOLITIS
Five percent of SLE patients are reported also to have obstructive physiology. Obliterative bronchiolitis has been documented in SLE, but is rare in contrast to rheumatoid arthritis. Organizing pneumonia, with inflammatory polyps protruding into bronchiolar lumens, is one of the interstitial patterns that occurs in acute lupus pneumonitis and in chronic interstitial lung disease in SLE, but this entity causes restriction rather than obstructive lung disease. Bronchiectasis may occur in up 20% of patients but is often asymptomatic. Large airway involvement including tracheal and subglottic stenosis, vocal fold paralysis, epiglottitis, and necrotizing tracheitis have all been reported, but are rare.
 RESPIRATORY MUSCLE DYSFUNCTION
RESPIRATORY MUSCLE DYSFUNCTION
It is estimated that weakness of the diaphragm and other respiratory muscles is found in 25% of patients with SLE. This accounts for the previously unexplained findings of dyspnea without evidence of interstitial or pulmonary vascular disease. These patients have subsegmental atelectasis, an elevated diaphragm on chest radiograph (Fig. 60-12), and restrictive physiology. This has been referred to as unexplained dyspnea and shrinking lungs syndrome. Although there is a reduction in static lung volumes, the diffusing capacity, when corrected for alveolar volume, remains normal, thereby distinguishing respiratory muscle dysfunction from interstitial lung disease. The likely explanation for this is a reduction in the transdiaphragmatic pressure generated during maximal inspiration, which in turn reduces static lung compliance, producing the linear atelectasis seen on the chest radiograph. Moreover, in the patients with respiratory muscle weakness, no evidence for a generalized neuromuscular disease can be found. The pathogenesis of respiratory muscle dysfunction remains unexplained, although phrenic nerve conduction abnormalities have been excluded.20 Abnormal diaphragmatic activation, due in part to voluntary inhibition due to pleuritic pain, may contribute to diaphragmatic dysfunction in this disorder. Clinical variables associated with the development of shrinking lung syndrome include: pleuritis, double-stranded DNA antibody and RNP antibody seropositivity, serositis, and a prolonged course of SLE.5 Corticosteroids are not a frequently effective treatment modality. Rituximab has been used successfully in the treatment of shrinking lung syndrome, albeit anecdotally in nature.21 Progression is uncommon and most patients stabilize. Positive-pressure ventilation, particularly at night, may improve these patients’ daytime symptoms, although there is limited evidence available. Rituximab has been used successfully in patients with respiratory muscle dysfunction with significant physiologic improvement.21
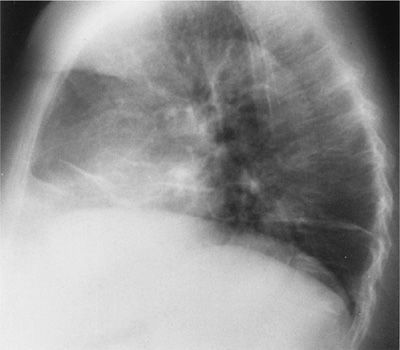
Figure 60-12 Diaphragmatic dysfunction in SLE. There is diaphragmatic elevation resulting in plate-like atelectasis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


