64 Pulmonary Hypertension and Thromboembolic Disease
Pulmonary Hypertension
Etiology and Pathogenesis
An expert committee of the World Health Organization has created a comprehensive diagnostic classification of pulmonary hypertension (Box 64-1). Of the many potential causes of pulmonary hypertension, the World Health Organization classification group 1—which includes idiopathic pulmonary artery hypertension (IPAH) and familial pulmonary artery hypertension (FPAH), diseases formerly referred to as “primary pulmonary hypertension”—merits special consideration. These diseases, characterized by proliferative and necrotic obliteration of the pulmonary microvasculature, are clinically indistinguishable except for family history. Both are devastating, despite advances in our understanding of their causes and treatment. Unlike the situation for many other causes of pulmonary hypertension, there are no reversible structural factors that can be addressed for individuals with IPAH and FPAH.
Box 64-1 2003 World Health Organization Classification of Pulmonary Hypertension
Adapted from Rubin LJ. Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based practice guidelines. Chest. 2004;126:7S–10S.
Clinical Presentation
Symptoms of pulmonary hypertension are common to multiple etiologies. Most patients with mild or moderate pulmonary hypertension are asymptomatic. Initial symptoms may be dyspnea with exertion, fatigue, and exertional intolerance. Many patients experience chest pain. Syncope suggests severe pulmonary hypertension with marked limitation of flow reserve. Hemoptysis is not common, but in some patients it is serious and fatal. Clinical presentation depends in part on the chronicity of the process. Adaptive changes in the right ventricle allow patients with chronic pulmonary hypertension to sustain near-systemic levels of pressures with minimal symptomatic effects. However, acute increases in pulmonary pressure, as with massive PE, cause immediate overt distress and, in many cases, collapse and death (Fig. 64-1).
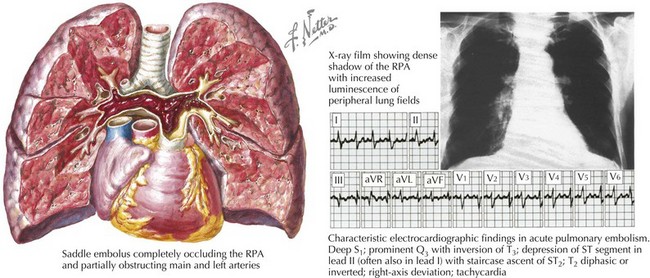
Two keys to the diagnosis of pulmonary hypertension are a high degree of suspicion raised by the clinical history and physical findings that suggest right ventricular (RV) failure and systemic congestion (see also Chapter 1). Increased PAPs are reflected in elevated RV systolic and, later, diastolic pressures. Because of chronically elevated RV systolic and diastolic pressure, the geometry of the right ventricle is altered, usually sufficiently to render the tricuspid valve incompetent. Tricuspid regurgitation creates a prominent v wave in the jugular venous pulse. Generally, the jugular venous pressure will be increased substantially in these patients, with filling of the deep neck veins above the clavicle visible with the patient sitting upright. Significant tricuspid regurgitation can also often be appreciated as pulsation of the liver. Less common and subtler physical findings with pulmonary hypertension are an RV precordial heave, an RV third heart sound, and increased intensity of the pulmonic component of the second heart sound.
Differential Diagnosis
Among numerous causes of pulmonary hypertension, the most common are chronic left ventricular dysfunction with or without valve disease and chronic lung diseases (see Box 64-1). These are usually recognized by history, and treatment is focused on the primary disease. All potential causes of secondary pulmonary hypertension should be excluded before a diagnosis of IPAH or FPAH is considered.
Diagnostic Approach
Table 64-1 lists the diagnostic tests and potential findings in the evaluation of patients with suspected pulmonary hypertension. Critical information on the degree and possible cause of pulmonary hypertension can be gained from a transthoracic echocardiogram. PAP can be estimated from the Doppler-derived velocity of tricuspid regurgitation and from the degree of RV dilation and hypertrophy. Echocardiography also provides data on left ventricular function, mitral valve structure and function, and the existence of an intracardiac shunt, all clues to the possibility that the pulmonary hypertension is an effect of cardiac disease.
Table 64-1 Evaluation of Patients with Suspected Pulmonary Hypertension
| Diagnostic Test | Potential Findings |
|---|---|
| Electrocardiography | |
| Chest radiography | |
| Echocardiography | |
| Pulmonary function testing with ABG | |
| Cardiac catheterization |
ABG, arterial blood gas; ASD, atrioventricular septal defect; COPD, chronic obstructive pulmonary disease; CT, computed tomography; PA, pulmonary artery; PAP, pulmonary artery pressure; LA, left atrial; LV, left ventricular; MRI, magnetic resonance imaging; RV, right ventricular; TR, tricuspid regurgitation; VSD, ventricular septal defect.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


