Vallerie V. McLaughlin, Marc Humbert
Pulmonary Hypertension
Pulmonary hypertension (PH) is defined as an increase in mean pulmonary arterial pressure (mPAP) of 25 mm Hg or greater at rest, as assessed by right-heart catheterization (RHC). PH has previously been called an orphan disease, that is, a condition that affects few individuals and is overlooked by the medical profession, health care systems, and pharmaceutical companies. Although rare, the concept that PH is overlooked cannot be considered to be the case today. Indeed, a number of recent important discoveries have improved our understanding of the disease, helped guide patient management, and laid foundations for future research. Since the mid-20th century, major achievements have been made in the field, from the development of RHC techniques to the first description of so-called primary PH and the progress achieved thanks to the National Institutes of Health (NIH) Primary Pulmonary Hypertension Registry and the World Pulmonary Hypertension conferences that have taken place five times in 40 years: 1973 (Geneva, Switzerland), 1998 (Evian, France), 2003 (Venice, Italy), 2008 (Dana Point, California, United States), and 2013 (Nice, France). The most recent guidelines provide a clear classification of the major clinical subcategories of PH (Table 74-1), among which pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH) have been subject to the most rapid advancement in terms of knowledge and treatment options in past decades.
TABLE 74-1
Updated Clinical Classification of Pulmonary Hypertension

ALK1 = activin receptor–like kinase type 1; BMPR2 = bone morphogenetic protein receptor type 2; COPD = chronic obstructive pulmonary disease; ILD, interstitial lung disease.
From Simonneau G, Catzoulis MA, Adatia I, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl):D34, 2013.
Definition
PH is a complex and multidisciplinary disorder. The term pulmonary hypertension refers to the presence of high pulmonary vascular pressure and can be the end result of a variety of different underlying disorders. By definition, PH is mPAP of 25 mm Hg or greater.1 The definition of normal versus abnormal is based on several factors: (1) the population resting mPAP is approximately 14 mm Hg, and 20 mm Hg encompasses 2 standard deviations above the mean; (2) a value of 25 mm Hg is therefore definitively above the normal distribution of values; and (3) the value of 25 mm Hg has, by consensus, been used to identify candidates for participation in clinical trials and registries. The definition for group 1, or PAH, also requires that left-sided cardiac filling pressure (pulmonary arterial wedge pressure [PAWP], left ventricular end-diastolic pressure [LVEDP], or left atrial pressure) be 15 mm Hg or less and that the calculated pulmonary vascular resistance (PVR) be 3 Wood units or greater.2
Anatomy
The lung has a unique double arterial blood supply from the pulmonary and bronchial arteries, as well as double venous drainage into the pulmonary and azygos veins. Each pulmonary artery accompanies the appropriate-generation bronchus and divides with it down to the level of the respiratory bronchiole. Pulmonary arteries are classified as elastic or muscular. Elastic arteries are conducting vessels that are highly distensible at low transmural pressure. As the arteries decrease in size, the number of elastic laminae decreases and smooth muscle increases. Eventually, in vessels between 100 and 500 µm, elastic tissue is lost from the media and the arteries become muscular. The intima of the pulmonary arteries consists of a single layer of endothelial cells and their basement membrane. The adventitia is composed of dense connective tissue in direct continuity with the peribronchial connective tissue sheath. The muscular arteries are 500 µm or smaller in diameter and are characterized by a muscular media bounded by internal and external elastic laminae. Arterioles are precapillary arteries smaller than 100 µm in outer diameter and composed solely of a thin intima and single elastic lamina. The alveolar capillaries are lined with a continuous layer of endothelium resting on a continuous basement membrane and focally connected to scattered pericytes located beneath the basement membrane. Within the respiratory units, the pulmonary arteries and arterioles are centrally located and give rise to precapillary arterioles, from which a network of capillaries radiates into the alveolar walls. The alveolar capillaries collect at the periphery of the acini and then drain into venules located in the interlobular and interlobar septa.
The bronchial circulation provides nutrition to the airways. The bronchial arteries ramify into a capillary network drained by bronchial veins; some empty into the pulmonary veins and the remainder into the systemic venous bed. The bronchial circulation therefore constitutes a physiologic right-to-left shunt. Normally, blood flow through this system amounts to approximately 1% of cardiac output (CO), and the resulting desaturation of left atrial blood is usually trivial.
Pathology
Different pathologic features characterize the diverse clinical PH groups. In PAH the pathologic lesions involve mainly the distal pulmonary arteries (<500 µm in diameter) and are characterized by medial hypertrophy, intimal proliferative and fibrotic changes (concentric, eccentric), adventitial thickening with moderate perivascular inflammatory infiltrates, complex lesions (plexiform, dilated lesions), and thrombotic lesions (Fig. 74-1). The pulmonary veins are classically unaffected in PAH, whereas in pulmonary venoocclusive disease (PVOD), the septal veins and preseptal venules are involved and exhibit occlusive fibrotic lesions, venous muscularization, patchy capillary proliferation, pulmonary edema, occult alveolar hemorrhage, lymphatic dilation with lymph node enlargement (vascular transformation of the sinus), and inflammatory infiltrates. In PVOD the distal pulmonary arteries are affected by medial hypertrophy, intimal fibrosis, and uncommon complex lesions.
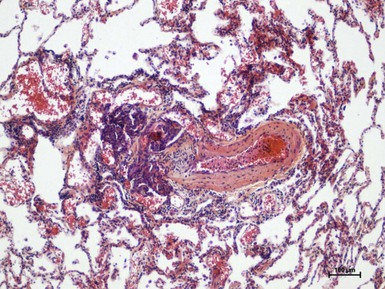
In PH caused by left-sided heart disease, the pathologic changes are characterized by enlarged and thickened pulmonary veins, pulmonary capillary dilation, interstitial edema, alveolar hemorrhage, and lymphatic vessel and lymph node enlargement. The distal pulmonary arteries may be affected by medial hypertrophy and intimal fibrosis. In PH caused by lung diseases and/or hypoxia, pathologic changes include medial hypertrophy and intimal obstructive proliferation of the distal pulmonary arteries. A variable degree of destruction of the vascular bed in emphysematous or fibrotic areas may also be present. In CTEPH, organized thrombi are tightly attached to the pulmonary arterial medial layer in the elastic pulmonary arteries and replace the normal intima. These thrombi may completely occlude the lumen or form different grades of stenosis, webs, and bands. In nonoccluded areas, a pulmonary arteriopathy indistinguishable from that of PAH (including plexiform lesions) may develop. Collateral vessels from the systemic circulation (from bronchial, costal, diaphragmatic, and coronary arteries) can grow and at least partially perfuse the areas distal to complete obstructions. In group 5 PH (Table 74-1), one can identify heterogeneous conditions with different pathologic bases for which the cause is unclear or multifactorial.
Pathobiology
PH has a multifactorial pathobiology in which an imbalance in vasoconstriction and vasodilation, thrombosis, and cell proliferation and remodeling of the walls of the pulmonary arteries contribute to increased PVR.3 As discussed earlier, pulmonary vascular remodeling involves the intima, media, and adventitia of small pulmonary arteries (diameter <500 µm), and all cell types (endothelial, smooth muscle, and fibroblast), as well as inflammatory cells and platelets, may play a significant role in the condition. Pulmonary vasoconstriction has been regarded as an early component of the PH process, and excessive vasoconstriction has been related to abnormal function or expression of potassium channels and to endothelial dysfunction. Endothelial dysfunction is characterized by impaired production of vasodilators such as nitric oxide (NO) and prostacyclin, along with overexpression of vasoconstrictors such as endothelin-1. Many of these abnormalities both elevate vascular tone and promote vascular remodeling and therefore represent logical pharmacologic targets. Recent genetic and pathophysiologic studies of PH have emphasized the relevance of several other mediators such as angiopoietins, serotonin, bone morphogenetic proteins (BMPs), and growth factors (platelet-derived growth factor [PDGF], fibroblast growth factor [FGF], epidermal growth factor [EGF], and the transforming growth factor-beta [TGF-β] superfamily). Abnormal proteolysis of the extracellular matrix, autoimmunity, and inflammation are also likely to contribute to the pathobiology of PH, and there is a growing literature on the role of cytokines and chemokines in pulmonary vascular remodeling.
Genetics
Idiopathic pulmonary arterial hypertension (IPAH) corresponds to sporadic disease without any family history of PAH or known triggering factor. In 1954, Dresdale and colleagues described the first case of familial PAH and demonstrated the existence of a heritable form of the disease. Since then, many cases of familial PAH have been described, and it was recognized that heritable/familial PAH is inherited as an autosomal dominant trait with incomplete penetrance (the disease will in turn develop in ≈20% of mutation carriers). A possible genetic anticipation phenomenon, characterized by age at onset of the disease significantly lower in each succeeding generation, was recently refuted for heritable/familial PAH.4 In 2000, BMPR2 (BMP receptor type 2) was identified as the first PAH-predisposing gene. This gene is located on the long arm of chromosome 2 (2q31-32) and encodes a type II receptor (BMPRII) belonging to the TGF-β receptor superfamily. The BMPRII receptor is involved in the regulation of growth, differentiation, and apoptosis of pulmonary artery endothelial and smooth muscle cells. When PAH occurs in a familial context, germline mutations in the BMPR2 gene are detected in approximately 70% to 80% of cases. BMPR2 mutations can also be detected in approximately 15% to 20% of apparently sporadic cases. The observation of a personal or familial history of hereditary hemorrhagic telangiectasia in patients with PAH allowed identification of other genes involved in the development of PAH, namely, activin A receptor type II–like kinase 1 (ACVRL1 or ALK1) and endoglin (ENG). Furthermore, mutations in other genes (i.e., BMPR1B, CAV1, and SMAD9) have been identified but are considerably less common. Of note, ALK1, ENG, and Smads proteins are all involved in the TGF-β signaling pathway. More recently, a novel channelopathy caused by mutation in the KCNK3 gene has been identified in familial and idiopathic cases of PAH, thus indicating for the first time that heritable disease may involve factors apparently independent of the TGF-β signaling pathway.5
Like IPAH, heritable/familial PAH affects twice as many females as males. It must be also emphasized that BMPR2 mutation carriers are younger at the time of diagnosis of PAH and have more severe hemodynamic compromise (higher mPAP, lower CO, lower PVR, and a lower likelihood of having an acute vasodilator component). Therefore BMPR2 mutation carriers are more likely to die sooner or to undergo transplantation than their IPAH counterparts are.6 It is currently recommended that genetic counseling be offered to family members of patients with heritable/familial PAH. These family members can be tested for the causal mutation (if any), and current research is attempting to identify the best PAH screening tool in asymptomatic mutation carriers. Currently, it is recommended that Doppler echocardiography be performed every 1 to 3 years or when signs and symptoms of PH develop in mutation carriers or first-degree relatives of those with heritable PAH. Other heritable forms of PH with a seemingly recessive mode of transmission have been described in patients with PVOD/pulmonary capillary hemangiomatosis (PCH). By using whole-exome sequencing it was found that recessive mutations in EIF2AK4 (also called GCN2) cosegregated with PVOD in all families studied in the French National Registry. Biallelic EIF2AK4 mutations were also detected in 5 of 20 histologically confirmed sporadic cases of PVOD/PCH. All mutations, either in a homozygous or compound-heterozygous state, disrupted function of the gene.7 EIF2AK4 encodes a serine-threonine kinase present in all eukaryotes that can induce changes in gene expression in response to amino acid deprivation. The pathophysiologic link between biallelic EIF2AK4 loss-of-function mutations and vascular cell proliferation and remodeling of lung vessels remains elusive.
Hemodynamics
The pulmonary circulation is characterized by high flow, low pressure, and low resistance. Normal mPAP at rest is 14.0 ± 3.3 mm Hg, and this value is independent of sex and ethnicity.8 Resting mPAP is just slightly influenced by age (<30 years, 12.8 ± 3.1 mm Hg; between 30 and 50 years, 12.9 ± 3.0 mm Hg; older than 50 years, 14.7 ± 4.0 mm Hg). Thus normal mPAP at rest is virtually independent of age and rarely exceeds 20 mm Hg. According to current guidelines, PH is defined as mPAP of 25 mm Hg or higher at rest, but more work is needed to better describe the natural history of patients with mPAP ranging from 21 to 24 mm Hg. PH can be classified as precapillary if PAWP is 15 mm Hg or lower or as postcapillary if PAWP is higher than 15 mm Hg. Some patients with PH may have a mixed picture characterized by elevated mPAP and PAWP with a transpulmonary gradient (mPAP − PAWP) higher than 12 mm Hg.
During exercise, mPAP is dependent on the exercise level and age. With mild exercise, mPAP is 19.4 ± 4.8 mm Hg in subjects younger than 50 years versus 29.4 ± 8.4 mm Hg in subjects 50 years or older. Exercise mPAP is related to age and frequently exceeds 30 mm Hg, especially in elderly individuals, which makes it difficult to define normal mPAP values during exercise. Given these circumstances, the diagnosis of exercise-induced PH was abandoned in 2008 because of insufficient evidence. Data have since shown that the upper limit of normal of mPAP flow relationships is 3 mm Hg/liter/min with a resistive vessel distensibility on the order of 1% to 2% change in diameter per mm Hg pressure and that higher pressure is associated with decreased exercise capacity. Exercise-induced PH is thus reemerging as a possible clinical entity with a physiologic substrate but remains a research topic until more is known about the natural history of this condition.
The normal pulmonary vascular bed offers less than 10% of the resistance to flow than does the systemic bed and can be approximated as the ratio of the drop in pressure (in mm Hg) to mean flow (in liter/min). PVR can be calculated as the ratio (mPAP − PAWP)/CO, whereas total pulmonary resistance (TPR) corresponds to the ratio mPAP/CO. The ratio can be multiplied by 80 to express the results in dyne-sec • cm−5 or be expressed in mm Hg/liter/min, which is referred to as a Wood unit. The calculated PVR in normal adults is 67 ± 23 dyne-sec • cm−5 (or 1 Wood unit). The physiologic range of PVR and TPR and the impact of exercise, age, and posture have been a matter of debate for many years. Supine resting PVR in subjects younger than 24, 24 to 50, 51 to 69, and 70 years or older is 61 ± 23, 69 ± 28, 86 ± 15, and 90 ± 39 dyne-sec • cm−5, respectively. Corresponding TPR is 165 ± 50, 164 ± 46, 226 ± 64, and 223 ± 45 dyne-sec • cm−5, respectively. During moderate exercise in subjects 50 years or younger, an 85% increase in CO is associated with a 25% decrease in TPR and a 12% decrease in PVR. At 51 to 69 years of age there is no significant decrease in TPR and PVR during exercise. In individuals 70 years or older, TPR may even increase by 17%, whereas PVR does not change significantly. At higher exercise levels, TPR decreases in all age groups.
Classification of Pulmonary Hypertension
The clinical classification of PH was most recently revised at the Fourth World Symposium on Pulmonary Hypertension held in Nice, France, in 20139 and is depicted in Table 74-1.
Group 1. Pulmonary Arterial Hypertension
Changes in classification have been made to reflect evolving understanding of the clinical and pathologic manifestations of PAH. PAH should not be considered a disease itself but is one measurable sign (elevated pulmonary arterial blood pressure) of an underlying pulmonary vasculopathy for which the clinic context must be appropriately diagnosed. Clinical experience and formal disease registry data bases make it increasing clear that the diseases grouped together within group 1 PAH, such as congenital heart disease (CHD) and connective tissue disease, have very different demographics, manifestations, and outcomes. The prevalence of group 1 PAH is in the range of 15 to 50 cases per million.
Etiology
Idiopathic Pulmonary Arterial Hypertension
Formerly referred to as primary pulmonary hypertension (PPH), IPAH is a rare disease of unknown cause and is the most common type of group 1 PAH in current-day registries. IPAH corresponds to a sporadic disease in which there is neither a family history of PAH nor an identified risk factor. It has a female preponderance (2:1 in the NIH registry, 4:1 in the current-day REVEAL registry). Even though the mean age at diagnosis was 37 in the NIH registry and approximately 50 in the more recent registries, IPAH can affect children and adults into their 70s.
Heritable Pulmonary Arterial Hypertension
Hereditary transmission of PAH has been reported in approximately 6% to 10% of patients with PAH. The genetic details of heritable PAH are discussed earlier.
Drug- and Toxin-Induced Pulmonary Arterial Hypertension
An association between anorexigens (appetite-suppressant drugs that increase release and block reuptake of serotonin) and PAH was initially observed in the 1960s when an epidemic of IPAH (then termed PPH) was noted in Europe after the introduction of aminorex fumarate. Structurally related compounds such as fenfluramine and dexfenfluramine were also demonstrated to be associated with the development of PAH in the 1980s and 1990s and have since been withdrawn from the market. Epidemiologic studies have also linked the development of PAH to rapeseed oil, l-tryptophan, and illicit drugs such as methamphetamines. More recently, the tyrosine kinase inhibitor dasatinib has been associated with the development of PAH.10 From the approval of dasatinib in November 2006 to September 30, 2010, nine incident cases of PAH in patients treated with dasatinib were identified in the French National Registry, which corresponds to an estimated incidence of 0.45% in patients exposed to dasatinib in France. Improvement is usually observed after cessation of the use of dasatinib.
Group 1′. Pulmonary Venoocclusive Disease and Pulmonary Capillary Hemangiomatosis
In rare instances, the typical histologic findings of PAH are associated with PVOD or PCH, a microvasculopathy. In addition to the histology of PAH, these entities also exhibit the findings of pulmonary venous hypertension, including pulmonary hemosiderosis, interstitial edema, and lymphatic dilation. Histologic proof is required for definitive diagnosis of PVOD and PCH, but surgical lung biopsy is a high-risk procedure in these patients and is therefore contraindicated. Although the risk factors and clinical features are usually indistinguishable from those of PAH, patients with PVOD may have crackles on examination and often have lower diffusing capacity of carbon monoxide and oxygen saturation at rest.19 High-resolution computed tomography (CT) of the chest in patients with PVOD is characterized by a higher frequency of centrilobular ground-glass opacities, septal lines, and mediastinal lymph node enlargement than in those with IPAH. The rapid development of pulmonary edema after the administration of PAH-specific therapy is sometimes the first clue to the appropriate diagnosis and can be life-threatening. Familial cases of PVOD/PCH have been described, often in consanguineous families. Recessive mutations in EIF2AK4 (also called GCN2) cosegregated with PVOD in 100% of familial and 25% of sporadic cases of histologically confirmed PVOD/PCH. These findings point to EIF2AK4 as the major gene that is linked to the development of PVOD/PCH and may be considered a possible future diagnostic tool in this rare condition.7 Survival of patients with PVOD is poor, and lung transplantation is the treatment of choice.
Clinical Diagnosis
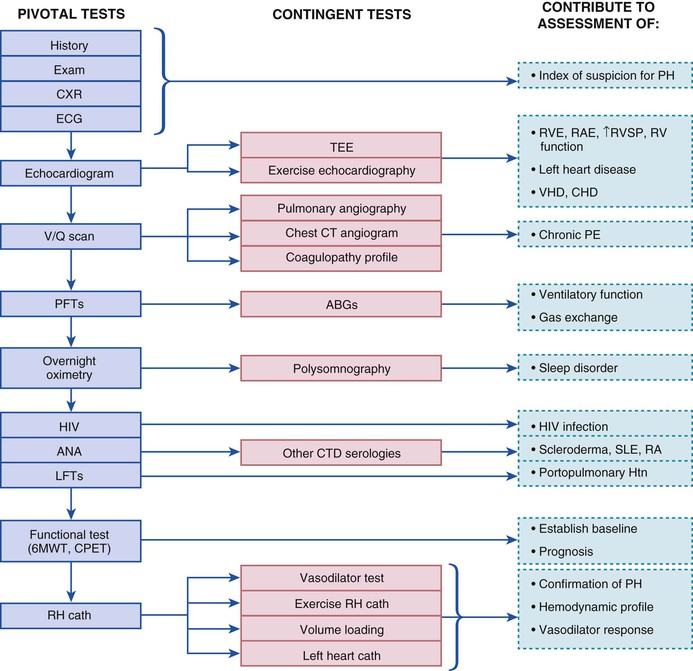
Given the multiple potential causes and factors contributing to the presence of PH, a methodical and extensive evaluation is warranted in most patients with common symptoms in whom the diagnosis is being considered (Fig. 74-2).
Symptoms
The most common initial symptoms of PH include exertional dyspnea or reduced exercise tolerance, chest pain, fatigue, and lightheadedness. Manifestations of more advanced disease include syncope, abdominal distention, and lower extremity edema attributable to right ventricular failure. Of course, the presence of risk factors for the development of PAH (e.g., connective tissue disease, family history, CHD, use of appetite suppressants) should heighten awareness of the disorder. In the NIH registry, the average time from the onset of symptoms to diagnosis was 2 years (see Classic References). Sadly, current-day registries suggest that delay in diagnosis persists. In the REVEAL registry, 21.1% of patients experienced symptoms for more than 2 years before PAH was recognized.20 Delay in diagnosis was most frequently observed in patients whose symptoms occurred at a younger age (<36 years) and in those with chronic obstructive pulmonary disease (COPD) or obstructive sleep apnea. It appears that young people in whom cardiopulmonary disease is considered less likely to be present or patients thought to have an alternative explanation for the symptoms are most at risk for delayed diagnosis.
Physical Examination
The physical examination can be subtle or nonspecific, but certain findings should raise suspicion for PAH. Features on the physical examination pertinent to the evaluation of PH are listed in Table 74-2.
TABLE 74-2
Features of the Physical Examination Pertinent to the Evaluation of Pulmonary Hypertension
| SIGN | IMPLICATION |
| Physical Signs That Reflect Severity of Pulmonary Hypertension | |
| Accentuated pulmonary component of S2 (audible at the apex in >90%) | High pulmonary pressure increases the force of pulmonic valve closure |
| Early systolic click | Sudden interruption of opening of the pulmonary valve into a high-pressure artery |
| Midsystolic ejection murmur | Turbulent transvalvular pulmonary outflow |
| Left parasternal lift | High right ventricular pressure and hypertrophy present |
| Right ventricular S4 (in 38%) | High right ventricular pressure and hypertrophy present |
| Increased jugular a wave | Poor right ventricular compliance |
| Physical Signs That Suggest Moderate to Severe Pulmonary Hypertension | |
| Moderate to severe PH | |
| Holosystolic murmur that increases with inspiration | Tricuspid regurgitation |
| Increased jugular v waves | |
| Pulsatile liver | |
| Diastolic murmur | Pulmonary regurgitation |
| Hepatojugular reflux | High central venous pressure |
| Advanced PH with right ventricular failure | |
| Right ventricular S3 (in 23%) | Right ventricular dysfunction |
| Distention of jugular veins | Right ventricular dysfunction, tricuspid regurgitation, or both |
| Hepatomegaly | Right ventricular dysfunction, tricuspid regurgitation, or both |
| Peripheral edema (in 32%) | |
| Ascites | |
| Low blood pressure, diminished pulse pressure, cool extremities | Reduced cardiac output, peripheral vasoconstriction |
| Physical Signs That Suggest a Possible Underlying Cause or Associations of Pulmonary Hypertension | |
| Central cyanosis | Abnormal ventilation-perfusion ratio, intrapulmonary shunt, hypoxemia, pulmonary-to-systemic shunt |
| Clubbing | CHD, pulmonary venopathy |
| Cardiac auscultatory findings, including systolic murmurs, diastolic murmurs, opening snap, and gallop | Congenital or acquired heart or valvular disease |
| Rales, dullness, or decreased breath sounds | Pulmonary congestion, effusion, or both |
| Fine rales, accessory muscle use, wheezing, protracted expiration, productive cough | Pulmonary parenchymal disease |
| Obesity, kyphoscoliosis, enlarged tonsils | Possible substrate for disordered ventilation |
| Sclerodactyly, arthritis, telangiectasia, Raynaud phenomenon, rash | Connective tissue disorder |
| Peripheral venous insufficiency or obstruction | Possible venous thrombosis |
| Venous stasis ulcers | Possible SCD |
| Pulmonary vascular bruits | Chronic thromboembolic PH |
| Splenomegaly, spider angiomas, palmar erythema, icterus, caput medusae, ascites | Portal hypertension |
CHD = congenital heart disease; SCD = sickle cell disease.
From McLaughlin VV, Archer SL, Badesch DB, et al: ACCF/AHA 2009 expert consensus document on pulmonary hypertension. A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 53:1573, 2009.
An accentuated pulmonic component of the second heart sound is present in most patients with PAH because of the high pulmonary pressure resulting in more forceful closure of the pulmonic valve. If a split S2 is audible at the apex, P2 may be accentuated and the possibility of PAH should be further investigated. The findings on physical examination are helpful to gauge the severity of PAH and to detect associated disorders as summarized in Table 74-2.
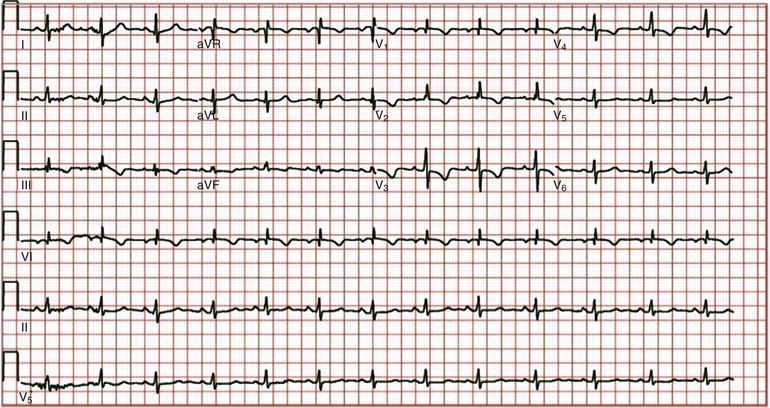
Electrocardiogram
Although the electrocardiogram is neither sensitive nor specific for PAH, it is an inexpensive, noninvasive test that can provide valuable information. Common electrocardiographic findings include right atrial enlargement, right-axis deviation, and right ventricular enlargement, often with a strain pattern (Fig. 74-3).
Chest Radiograph
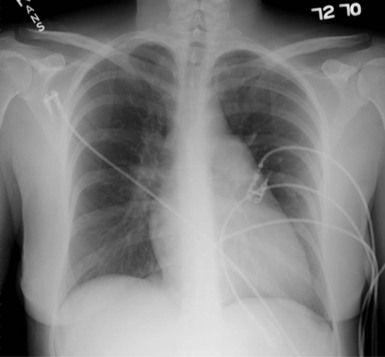
Findings on the chest radiograph that suggest the presence of PH include enlarged main and hilar pulmonary artery shadows with “pruning” or attenuation of the peripheral vasculature (Fig. 74-4) and right ventricular enlargement, which is best appreciated on the lateral view. Other findings on the chest radiograph may point to an associated diagnosis, such as hyperinflation with flat diaphragms (COPD) or pulmonary venous congestion (left-sided heart disease).
Echocardiogram
If PH is suspected from the history, assessment of risk factors, and physical examination, an echocardiogram is the next appropriate study (see Chapter 14). Echocardiography also serves as a useful noninvasive screening test for PH in at-risk populations (e.g., scleroderma, CHD). Doppler echocardiography can simultaneously provide an estimate of right ventricular systolic pressure and the functional and morphologic sequelae of PH and give clues to other potential cardiac causes of PH (see Fig. 14-78). Common echocardiographic features of PAH include right atrial enlargement, right ventricular enlargement and dysfunction, small underfilled left-sided heart chambers, interventricular septal flattening, tricuspid regurgitation with elevated velocity, and reduced tricuspid annular plane systolic excursion (TAPSE) (see Fig. 14-21). A saline contrast injection can be used to detect an intracardiac shunt. One must acknowledge the limitations of the estimated right ventricular systolic pressure because of multiple potential sources of error in this measurement. In any given patient, the estimated right ventricular systolic pressure must be put into context with the patient’s symptoms, previous medical history, and other findings on the two-dimensional echocardiogram. In the absence of other potential causes of PH, such as left-sided heart disease or hypoxemic lung disease, an estimated right ventricular systolic pressure greater than 40 mm Hg generally warrants further evaluation in a patient with unexplained dyspnea. Other echocardiographic findings that warrant further evaluation include right atrial and right ventricular enlargement and abnormal interventricular septal motion. Guidelines for echocardiographic assessment of the right heart in adults have recently been published.21
The echocardiogram often provides information on the possibility of group 2, or PH caused by left-sided heart disease. Left ventricular systolic or diastolic dysfunction and aortic and mitral valvular heart disease are easily assessed on an echocardiogram. The presence of left atrial enlargement suggests chronically elevated left-sided filling pressure.
In some instances, particularly in the assessment of CHD, a transesophageal echocardiogram provides additional information. The role of exercise echocardiography is controversial at this time.
The most important echocardiographic prognostic indicators for PAH include the presence of pericardial effusion and the severity of right ventricular dysfunction. Estimated right ventricular systolic pressure is less meaningful prognostically, and in fact this value may fall as the disease progresses and the right ventricle becomes more dysfunctional.
Right-Heart Catheterization
Invasive hemodynamic assessment by RHC is pivotal in the evaluation of any patient with suspected PAH. RHC is typically performed after the noninvasive testing for PH described earlier. Some patients initially suspected of having PAH will not require RHC because they have had an alternative diagnosis established by noninvasive testing. However, all patients who are still suspected of having PAH after noninvasive evaluation should undergo RHC before the initiation of therapy. The usefulness of RHC is dependent on the accuracy and completeness of the data obtained. Essential measurements during RHC include the following:
• Oxygen saturation (superior and inferior vena cavae, pulmonary and systemic arteries)
• Left-sided filling pressure (PAWP, left atrial pressure, or LVEDP)
• PVR
Misinterpretation of PAWP is a common pitfall in the invasive diagnosis of PH. PAWP should be measured at end-expiration and in several different segments of the pulmonary vasculature. LVEDP should be determined if there is any doubt about the accuracy of the PAWP tracing or if the results are unexpected in a given patient. A fluid challenge may be necessary to elicit the presence of diastolic dysfunction.
Acute vasodilator testing should be performed in most patients with IPAH. Exceptions include patients who would not be candidates for long-term therapy with a calcium channel–blocking agent, such as those with hemodynamic instability or overt right-sided heart failure. Responders are rare among patients with associated PAH. The most common agents used for acute vasodilator testing are inhaled NO, intravenous epoprostenol, and intravenous adenosine. An acute response is defined as a decrease in mPAP by at least 10 mm Hg to an absolute mPAP value lower than 40 mm Hg in the setting of unchanged or increased CO.2,28
Compliance with Guidelines
Sadly, despite publication of the diagnostic algorithmic recommendations in multiple sources, many patients are treated with PAH-specific therapies without having completed the required diagnostic studies. A recent initiative studied compliance with the American College of Chest Physicians diagnostic algorithm.29 This initiative demonstrated that compliance with the guidelines was poor and that the most frequent studies that were not performed included the ventilation-perfusion scan (57%), HIV serology (29%), and connective tissue disease serology (50%). Ten percent of patients were assigned the diagnosis of PAH without RHC. Only 7% of patients being treated with calcium channel–blocking agents fulfilled the criteria for an acute responder. Tools to improve compliance with the guidelines may improve care and outcomes in patients with PAH. Establishing a correct diagnosis is critical before the commencement of PAH-specific therapy.
Treatment
Treatment of PAH has evolved considerably over the past decade, in part because of advances in knowledge of the disease and the availability of agents that target known derangements in the pathobiologic process. Multiple treatment algorithms have been published over the recent years. The algorithm from the 2013 World Symposium in Nice, France,30 is reproduced in Figure 74-5. Treatment decisions are often made with the severity of illness in mind. Table 74-3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


