Pulmonary Atresia and Intact Ventricular Septum
David G. Nykanen
Robert Mark Freedom 1941–2005
A discussion of pulmonary atresia and intact ventricular septum would be incomplete without acknowledging the legacy of Robert M. Freedom. As a clinical cardiac morphologist his monogram described the diversity of this malformation, emphasizing the complexity of a disorder that appears so simple on the surface (1). His understanding of the nature of this condition has endured the test of time and continues to influence our treatment strategies today. To those who were fortunate to know him, he was a passionate, tireless teacher dedicated to the concept that the practice of knowledge-based medicine incorporates observations gained from both an understanding of the literature and the recognition of the value of cumulative experience. Scholars of pediatric cardiology will continue to identify Dr. Robert Freedom’s contributions to our understanding of pulmonary atresia and intact ventricular for decades to come. It is appropriate that many of his insights continue to be reflected in the pages that follow.
Pulmonary atresia and intact ventricular septum was first described in 1783 by Hunter (2) and then revisited 86 years later in 1869 by Peacock (3). While its name focuses primarily on membranous or muscular atresia of the right ventricular outflow tract, this disorder is characterized by striking heterogeneity of the right ventricle, its inlet, and its functional size. Furthermore, many of these patients have connections between the right ventricle and subepicardial coronary arteries and a predisposition for an unusual coronary circulation. These connections were originally a pathologic curiosity; however, management can be dictated by their nature. Prognosis appears to relate to the nature of the coronary circulation in the patient with a right ventricle at high pressure, or alternatively to the presence of severe tricuspid regurgitation in the setting of a low-pressure right ventricle (4,5,6). It is truly a complex, diverse disorder that challenges surgical and catheter-based interventional strategies. Current interventional algorithms range from achieving a biventricular circulation through variations of the cavopulmonary circulation to cardiac transplantation.
Epidemiology
Data obtained from the New England Regional Infant Cardiac Program identified 75 patients with this disorder, accounting for 3.1% of all infants enrolled in the study. The Baltimore–Washington Infant Study defined the prevalence for this disorder as 0.083/1,000 live births (7). Despite being uncommon, when integrated into the overall statistics of congenital heart disease, pulmonary atresia and intact ventricular septum is one of the more common types of cyanotic congenital heart disease in the neonate, along with transposition of the great arteries and pulmonary atresia with ventricular septal defect. A published study from the United Kingdom and Eire defined the incidence of pulmonary atresia and intact ventricular septum to be 4.1/100,000 live births (8). Overall, by the best estimates pulmonary atresia and intact ventricular septum occurs at a rate of 0.6/10,000 live births. However, if one includes pregnancies that are spontaneously or electively aborted after 20 weeks gestational age with live births the rate has been reported as much as 10 times higher at 0.6/1,000.
Fetal echocardiography provides a unique window to study the later phases of the fetal cardiac development recognizing that cardiac organogenesis is complete by about 8 weeks of gestation. There is increasing evidence that fetuses with severe tricuspid regurgitation may not fare well. Such fetuses are known to develop right-sided heart failure with pleural and pericardial effusions, ascites, pulmonary hypoplasia, and fetal death. Thus, fetal loss might be anticipated in a specific subset of patients with pulmonary atresia, intact ventricular septum, extremely severe tricuspid regurgitation, and a low-pressure right ventricle. The data collected from the United Kingdom and Eire also showed that termination of pregnancy once this diagnosis was established led to an important reduction in live-born incidence in mainland Britain (8). Prenatal echocardiography is becoming an important predictor of postnatal management of this patient population and will likely grow to include strategic application of in utero intervention (9,10,11,12,13).
Definition
The usual form of pulmonary atresia and intact ventricular septum occurs in a left-sided heart with usual atrial arrangement, concordant atrioventricular connections, and concordant ventriculoarterial connections. As its name implies, the right ventricular outflow tract is imperforate and this can be either membranous or represented by a longer segment muscular atresia. The ventricular septum is functionally intact. A patent arterial duct usually provides pulmonary flow. Very rarely multiple direct aortopulmonary collaterals originating from the descending thoracic aorta are the sole sources of pulmonary arterial supply. Nonconfluent pulmonary arteries, each supplied by a separate patent arterial duct, also have been recognized, but this situation is also uncommon (14).
Morphogenesis
Kusche and Van Mierop (15) suggested that pulmonary atresia with ventricular septal defect occurs earlier in cardiac morphogenesis than pulmonary atresia and intact ventricular septum. This conclusion is based on an analysis of a number of morphologic factors, including the diameter of the pulmonary trunk, the morphology of the pulmonary valve, and the morphology and topography of the ductus arteriosus. They postulated that pulmonary atresia and ventricular septal defect occurs early in cardiac morphogenesis at or shortly after partitioning of the truncoconal part of the heart
but before closure of the ventricular septum. Conversely, they suggest that pulmonary atresia and intact ventricular septum probably occurs after cardiac septation. For some forms of hearts with pulmonary atresia and intact ventricular septum, it is possible that their conclusions about the timing of maturational arrest are correct; specifically for patients with pulmonary atresia and intact ventricular septum, a nearly normal-sized right ventricle, and an imperforate “tricuspid” pulmonary valve whose commissures are completely fused.
but before closure of the ventricular septum. Conversely, they suggest that pulmonary atresia and intact ventricular septum probably occurs after cardiac septation. For some forms of hearts with pulmonary atresia and intact ventricular septum, it is possible that their conclusions about the timing of maturational arrest are correct; specifically for patients with pulmonary atresia and intact ventricular septum, a nearly normal-sized right ventricle, and an imperforate “tricuspid” pulmonary valve whose commissures are completely fused.
Embryologically this defect must occur after ventricular septation is complete as the septum is by definition intact. An abnormality of intracardiac blood flow may result in atresia of the valve thus further altering blood flow and ventricular growth. Animal experiments have demonstrated that reduced flow to a cardiac structure, even late in gestation, can have devastating effects on the development of associated structures (16,17). The concept is further evidenced by current (as yet unproven) experimental fetal interventional strategies for treatment during pregnancies that are aimed at restoring normal flow patterns in an effort to promote growth or prevent regression of cardiac chamber size. While human fetuses with pulmonary atresia and intact ventricular septum demonstrate abnormalities in intracardiac flow, the specific etiology of the flow abnormality remains unknown.
The tricuspid valve in this condition is usually abnormal. This makes intuitive sense as any blood entering the fetal right ventricle has no egress. Hence it must regurgitate or develop an alternate means of decompression such as through connections with the coronary circulation. The annulus may become hypoplastic, the leaflets of the valve abnormal and the chords supporting the valve malformed or damaged by high pressures in the ventricle. Once the valve begins to leak its structure is further altered by turbulent flow.
Another very important prognostic feature concerns the presence or absence of ventriculocoronary connections and coronary artery abnormalities (18). Conceptually if the pressure is very high in the right ventricle and the blood has nowhere to go, the ventricle can develop these connections to partially decompress in fetal life. Once these connections have formed they have not been demonstrated to disappear. Shear forces created by blood flowing at high pressure within these connections are thought to underpin the progressive stenosis and interruption of the coronary arteries that can effectively render the circulation of major portions of heart muscle dependent on flow from the right ventricle. This is recognized after birth as a “right ventricular–dependent coronary circulation” and in its worst form is a contraindication to decompressing the right ventricle by surgery or transcatheter methods.
The pulmonary valve is derived from endocardial tissue within the conotruncus. Abnormalities in morphology of the pulmonary valve can result from failure of normal development of the valve itself. In this setting the valve consists of the usual three leaflets but they have markedly thickened abnormal cusps of myxomatous tissue. It is often accompanied by abnormalities of the valve annulus and the sinotubular junction. These types of valve abnormalities have been linked to genetic conditions such as Noonan syndrome and Williams syndrome as well as environmental exposures such as rubella. Here the problem clearly occurs early as a result of abnormal formation of the valve itself. This is in contrast to the valve that has been subject to abnormalities of flow after it has developed where the leaflets are thickened and fused together with a near-normal appearing sinotubular junction. It is important to make the distinction between an imperforate valve (membranous atresia) and atresia of the entire outflow tract (muscular atresia) as the latter predicts a condition that is more likely to have extensive ventriculocoronary connections, coronary ostial stenosis, and a poorer outcome (19).
Pathology and Physiology
Gross External Inspection
The heart may be only mildly enlarged, or it may be massively enlarged, with a hugely dilated right atrium occupying much of the right hemithorax. In this latter situation, the lungs may be compressed by the enlarged heart and may exhibit a varying degree of hypoplasia. When the heart is only mildly enlarged, the course of the anterior descending coronary artery in the anterior interventricular sulcus outlines a smaller than normal right ventricle. The right atrium is usually somewhat enlarged, accounting for the cardiomegaly. The right ventricle may be profoundly thinned and this may be apparent even with the heart in situ.
From external inspection of the heart, there may be obvious clues that there are significant abnormalities of the coronary artery circulation. The coronary arteries may be obviously thickened and nodular, and rarely the coronary arteries may be seen connecting with the pulmonary trunk. Severe abnormalities may be readily apparent with epicardial aneurysmal dilation (Fig. 40.1). So-called dimples may be observed on the epicardial surface of the heart, usually, but not exclusively, in association with the subepicardial coronary arteries. Such dimples may be considered the external stigmata of ventriculocoronary connections and may indicate the site of such connections.
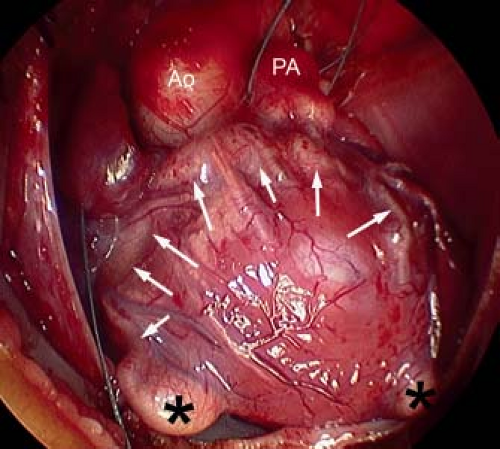 Figure 40.1 Intraoperative photograph of the epicardial surface of the heart of a patient with pulmonary atresia and intact ventricular septum with right ventricle to coronary connections. The coronary arteries are dilated (arrows) with aneurysmal dilation (asterisks). Ao, Aorta; PA, Pulmonary artery. |
Nature of the Pulmonary Atresia
Braunlin et al. (20) documented the morphologic bases for pulmonary atresia in this disorder, correlating the type of imperforate pulmonary valve with the character of the right ventricle and its infundibulum. In patients with a well-formed infundibulum the imperforate pulmonary valve exhibits three semilunar cusps with complete fusion of the commissures (Fig. 40.2). The pulmonary valve is primitive in patients with a diminutive right ventricle and a severely narrowed or atretic infundibulum. As described in the
previous section, the muscular nature of the infundibulum has received morphologic, clinical, and angiocardiographic attention, and the nature of those right ventricular muscle bundles contributing to the infundibular atresia has been described.
previous section, the muscular nature of the infundibulum has received morphologic, clinical, and angiocardiographic attention, and the nature of those right ventricular muscle bundles contributing to the infundibular atresia has been described.
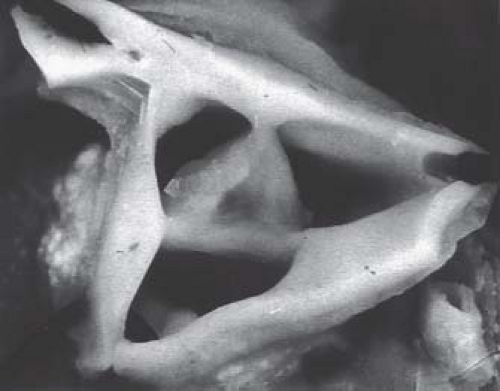 Figure 40.2 Appearance of pulmonary valve in a patient with pulmonary atresia and intact ventricular septum. The valve leaflets are reasonably well identified. |
Great Veins, Atrial Septum, Coronary Sinus, and Venous Valves
A peculiar relationship exists between persistent right venous valve, ventriculocoronary connections, and pulmonary atresia with intact ventricular septum. It would be too simplistic, indeed incorrect, to speculate that a persistent venous valve is causal to right heart hypoplasia. The coronary sinus usually terminates in the right atrium. Stenosis and atresia of the coronary sinus ostium have been observed, with decompression through an unroofed coronary sinus–left atrium fenestration.
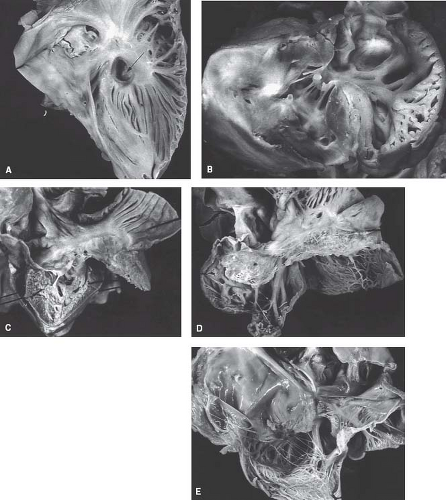 Figure 40.3 Spectrum of tricuspid valve anomalies in pulmonary atresia and intact ventricular septum. A: Severely stenotic tricuspid orifice (arrow) viewed from the right atrium. B: Stenotic and hypoplastic tricuspid valve with a very muscular annulus. C: Nearly normal tricuspid valve in a patient with a normal-sized right ventricle. D: Severely dysplastic tricuspid valve in a newborn with large right ventricle and severe tricuspid regurgitation; the right ventricle is very thinned. E: Profound Ebstein anomaly of the tricuspid valve in a newborn with large right ventricle and severe tricuspid regurgitation. |
Because of the obligatory right-to-left shunt at atrial level, with rare exception, there is either a patent foramen ovale or true secundum atrial septal defect. Premature closure of the foramen has been observed in this disorder, usually with fetal death. Rarely, if the interatrial septum is intact or nearly so, alternative pathways for systemic venous return have been recognized, including coronary sinus–left atrial fenestration. The septum primum may assume aneurysmal proportions in patients with a restrictive atrial septal defect, and its herniation through or obstruction of the left ventricular inflow has been observed.
Tricuspid Valve
The tricuspid valve is rarely normal in patients with pulmonary atresia and intact ventricular septum. This atrioventricular valve demonstrates the continuum of abnormalities and a functional impact that ranges from extreme stenosis to profound regurgitation (21) (Fig. 40.3). The stenotic valve can demonstrate a hypoplastic obstructive annulus that may be muscularized with a very abnormal valve apparatus consisting of a thickened free valve margin, shortened dysplastic chordae, and papillary muscle abnormalities. The severely regurgitant valve can be characterized by a dilated annulus. In this situation, the valve exhibits both profound displacement with the severest form of Ebstein anomaly and dysplasia. In some severely regurgitant valves, the valve is not displaced, but it is extremely dysplastic. Rarely, the valvar orifice may be virtually unguarded, a situation similar to profound Ebstein anomaly (22). The most severely stenotic and obstructive tricuspid valve is observed in patients with the most hypoplastic of right ventricles. Conversely, patients with a large right ventricle usually have severe tricuspid regurgitation with a valve exhibiting features of Ebstein anomaly and dysplasia. This latter malformation represents a major management challenge with a poor overall prognosis.
Right Ventricle
Clinicians and pathologists have attempted to characterize the size of the right ventricle in this disorder for decades. Methodologies have included angiocardiographic volumetric analysis, a variety of measurements of the inlet/outlet axis and the convention advocated
by the Congenital Heart Surgeons Study (CHSS); the use of the so-called tricuspid diameter Z-value (5). This reflects the deviation of the valvar diameter from that expected for body surface area and was based on an early pathologic study of the normal quantitative anatomy. Data from the CHSS showed that the Z-value of the tricuspid valve was correlated with the size of the right ventricular cavity (r = 0.68, p <0.0001) and this relationship has been confirmed in more recent studies as well (23). This continues to be referenced to the present day.
by the Congenital Heart Surgeons Study (CHSS); the use of the so-called tricuspid diameter Z-value (5). This reflects the deviation of the valvar diameter from that expected for body surface area and was based on an early pathologic study of the normal quantitative anatomy. Data from the CHSS showed that the Z-value of the tricuspid valve was correlated with the size of the right ventricular cavity (r = 0.68, p <0.0001) and this relationship has been confirmed in more recent studies as well (23). This continues to be referenced to the present day.
Others have advocated a semimorphologic approach to the right ventricle. Although there is no consensus as to whether the morphologically right ventricle is embryologically a bipartite or tripartite structure, there are examples of congenitally malformed hearts that may support the view of the right ventricle as a tripartite structure. Taking this latter approach, the normal right ventricle can be considered to be composed of confluent inlet, apical trabecular, and outlet components. This approach has been used widely in the categorization of patients with pulmonary atresia and intact ventricular septum. Thus, some patients have pulmonary atresia and intact ventricular septum wherein the right ventricle is particularly well formed and all three components are well represented. In others, the right ventricle is extremely underdeveloped and seemingly limited to an inlet only (24) (Figs. 40.3C–E and 40.4). Attempts to characterize the size of the right ventricle are plagued by difficulty in assessing the contribution of muscular hypertrophy of the apical and outlet myocardium (25). This has tremendous impact on clinical management, as it appears that relief of outflow tract obstruction with associated pulmonary insufficiency may result in remodeling with regression of hypertrophy that can result in real or perceived growth of the right ventricle (25,26,27,28,29,30,31,32).
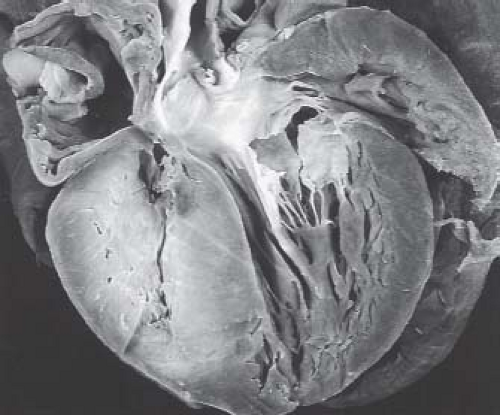 Figure 40.4 Severe right ventricular underdevelopment in a patient with pulmonary atresia and intact ventricular septum. Severely hypoplastic right ventricle with free-wall and septal hypertrophy. |
Left Atrium, Left Ventricle, and Aortic Valve
The left atrium usually receives the pulmonary veins in a normal fashion, although one or more pulmonary veins may connect anomalously to the systemic circulation. The left ventricle may
exhibit variable degrees of hypertrophy, especially in patients who survive past infancy. Some years ago, attention was given to a convexity of the outlet portion of the interventricular septum that occurred in those patients with small and extremely hypertensive right ventricles (Fig. 40.5). In this setting, severe left ventricular outflow tract obstruction resulting in death occurring after surgical creation of a cavopulmonary connection has been observed when there is an unfavorable change in the ratio between left ventricular mass and end-diastolic volume (33,34). Aortic valve stenosis has been described in patients with pulmonary atresia and intact ventricular septum, including the neonate with critical aortic stenosis and the somewhat older child with severe aortic valve stenosis (35). With rare exceptions, the aortic arch is left sided.
exhibit variable degrees of hypertrophy, especially in patients who survive past infancy. Some years ago, attention was given to a convexity of the outlet portion of the interventricular septum that occurred in those patients with small and extremely hypertensive right ventricles (Fig. 40.5). In this setting, severe left ventricular outflow tract obstruction resulting in death occurring after surgical creation of a cavopulmonary connection has been observed when there is an unfavorable change in the ratio between left ventricular mass and end-diastolic volume (33,34). Aortic valve stenosis has been described in patients with pulmonary atresia and intact ventricular septum, including the neonate with critical aortic stenosis and the somewhat older child with severe aortic valve stenosis (35). With rare exceptions, the aortic arch is left sided.
Pulmonary Circulation
Confluent pulmonary arteries usually receive flow from a left-sided ductus arteriosus. Rarely, nonconfluent pulmonary arteries are supported by bilateral ductus arteriosus or aortopulmonary collaterals (36). A main pulmonary trunk is almost always present. Unlike the patient with pulmonary atresia with ventricular septal defect, the caliber of the pulmonary arteries in patients with pulmonary atresia and intact ventricular septum is rarely a major determinant of outcome. This may be in part mediated by the observation that the arterial duct tends to close earlier in patients with pulmonary atresia and intact septum than it does in patients with pulmonary atresia and ventricular septal defect (37). More recently transcatheter means of maintaining pulmonary blood flow such as stenting of the arterial duct have necessitated increased awareness of the assessment the neonatal anatomic details of the pulmonary artery at the insertion of the patent ductus arteriosus (38,39,40,41).
Myocardial Abnormalities
The myocardium of patients with pulmonary atresia and intact ventricular septum can demonstrate a wide range of abnormalities. Ischemia, fibrosis, infarction, and myocardial rupture have been observed in these patients (42,43,44,45,46,47) (Fig. 40.6). Other abnormalities include myocardial disarray, the appearance of the so-called spongy myocardium, and ventricular endocardial fibroelastosis. In this regard, an inverse relationship exists between ventricular endocardial fibroelastosis and extensive ventriculocoronary communications. Perhaps this observation is fundamental to the frequent finding of ventriculocoronary connections in pulmonary atresia and intact ventricular septum: dense, “sugar-coated” right ventricular endocardial sclerosis is uncommon. Conversely, dense, sugar-coated left ventricular endocardial sclerosis is common in patients with a hypoplastic left heart syndrome; but a perforate mitral valve, and thus ventriculocoronary connections, is uncommon. The right ventricular myocardium may be particularly thinned in babies with severe tricuspid regurgitation.
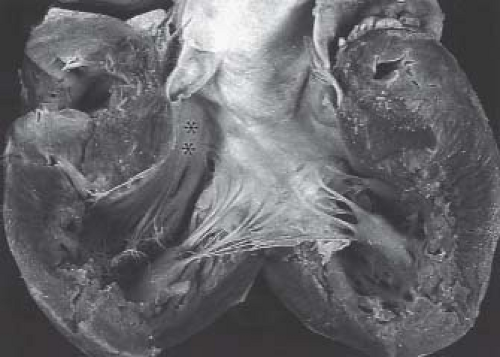 Figure 40.5 Note septal convexity (asterisks) in a patient with pulmonary atresia and intact ventricular septum. It has the potential for promoting left ventricular outflow tract obstruction after Fontan repair. |
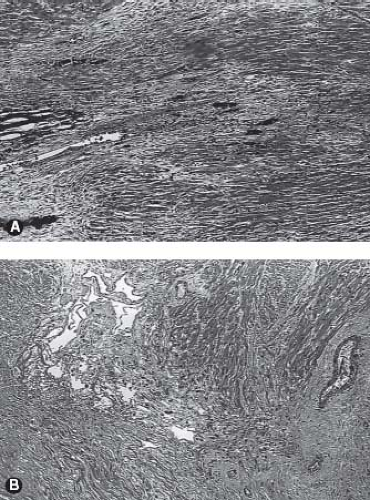 Figure 40.6 Myocardial abnormalities seen in some patients with pulmonary atresia and intact ventricular septum. A: Acute myocardial infarction with rich inflammatory infiltrate and loss of cellular integrity. B: So-called spongy myocardium with endothelial-lined vascular lakes. |
Coronary Arteries
It is important to gain an understanding of the status of the coronary circulation in patients with pulmonary atresia and intact ventricular septum prior to proceeding with an interventional algorithm as myocardial ischemia may be related to the presence and extent of these ventriculocoronary connections. An extensive literature has documented the vast array of changes in the coronary arteries among some patients with pulmonary atresia and intact ventricular septum (48,49,50,51,52). Several articles have characterized the histopathologic alterations of the involved coronary arteries. This process is not characterized by inflammation, as once thought, but more appropriately as myointimal hyperplasia with a rich background of glycosaminoglycans. There is a wide spectrum of histopathologic lesions of both the extramural and intramural coronary arteries. These lesions range from mild degrees of intimal and medial thickening in which a continuous internal elastic lamina and normal lumen are present to a loss of normal arterial wall morphology with replacement of
the arterial wall by fibrocellular tissue containing irregular, disorganized elastin strands and severe stenosis or obliteration of the arterial lumen. Some have designated these changes as fibroelastosis of the coronary arteries, but emphasis would be more appropriately focused on myointimal hyperplasia. Staining for glycosaminoglycans shows the prominence of ground substance formation by the activated smooth-muscle cells rather than the reduplicated elastic and collagen characteristic of fibroelastosis (Fig. 40.7). This pathologic process leads to a profound distortion of the normal architecture, resulting in endothelial irregularity, stenosis, or interruption. Such coronary arterial involvement occurs only in patients with ventriculocoronary connections and, by inference, with a hypertensive right ventricle. The pathogenesis of these arterial lesions is likely predicated on the repeated and sustained injury to the coronary arterial intima from high-pressure right ventricular systolic turbulent flow mediated by the presence of the ventriculocoronary connections. Intramural or extramural coronary arteries remote from the ventriculocoronary connections do not demonstrate these arterial lesions, nor do the coronary arteries of hearts without ventriculocoronary connections. These lesions have been found in fetal hearts with pulmonary atresia and intact ventricular septum and in hearts of the immediate newborn. Ventriculocoronary connections do not occur in thin-walled, low-pressure right ventricles.
the arterial wall by fibrocellular tissue containing irregular, disorganized elastin strands and severe stenosis or obliteration of the arterial lumen. Some have designated these changes as fibroelastosis of the coronary arteries, but emphasis would be more appropriately focused on myointimal hyperplasia. Staining for glycosaminoglycans shows the prominence of ground substance formation by the activated smooth-muscle cells rather than the reduplicated elastic and collagen characteristic of fibroelastosis (Fig. 40.7). This pathologic process leads to a profound distortion of the normal architecture, resulting in endothelial irregularity, stenosis, or interruption. Such coronary arterial involvement occurs only in patients with ventriculocoronary connections and, by inference, with a hypertensive right ventricle. The pathogenesis of these arterial lesions is likely predicated on the repeated and sustained injury to the coronary arterial intima from high-pressure right ventricular systolic turbulent flow mediated by the presence of the ventriculocoronary connections. Intramural or extramural coronary arteries remote from the ventriculocoronary connections do not demonstrate these arterial lesions, nor do the coronary arteries of hearts without ventriculocoronary connections. These lesions have been found in fetal hearts with pulmonary atresia and intact ventricular septum and in hearts of the immediate newborn. Ventriculocoronary connections do not occur in thin-walled, low-pressure right ventricles.
The coronary abnormalities in patients with pulmonary atresia and intact ventricular septum embrace the same spectrum of abnormalities as those seen in patients with otherwise normal hearts, including abnormalities of origin, epicardial course, and number. A single coronary artery may originate from the aorta or, rarely, from the pulmonary trunk. Several congenital and acquired conditions of the coronary circulation are specific to pulmonary atresia and intact ventricular septum and impact surgical management. These conditions include an absence of a proximal aortocoronary connection between one or both coronary arteries, coronary arterial stenosis or interruption, or a so-called coronary–cameral fistula with a major fistula between right or left coronary artery and the right ventricle. Particularly rare arterial connections such as those from the descending thoracic aorta or the gastric artery to the coronary circulation have been described.
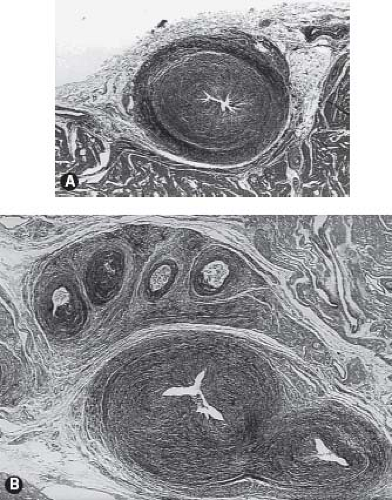 Figure 40.7 Spectrum of severe coronary arterial lesions in infants with pulmonary atresia and intact ventricular septum. A: Virtual obliteration of coronary arterial lumen by a rich cellular process. B: Obliteration of extramural coronary arteries in a different patient with a right ventricular–dependent coronary circulation. |
Right Ventricular–Dependent Coronary Artery Circulation
Intrinsic to awareness of ventriculocoronary connections in this disorder and their impact on the myocardium is the concept of a right ventricular–dependent coronary circulation (Table 40.1). In the normal circulation, it is in large part the aortic diastolic pressure that is the driving pressure for coronary flow. Factors that reduce aortic diastolic pressure or shorten diastole will compromise coronary flow. The presence of ventriculocoronary artery connections may promote coronary artery stenosis and interruption, and aortic diastolic pressure may not be sufficient to drive coronary blood flow when obstructive lesions are present within the coronary circulation. It is important to remember that these infants are hemodynamically fragile, tachycardic, and often receiving prostaglandin or palliated with a systemic-to-pulmonary artery shunt to augment pulmonary flow. Notably these therapeutic maneuvers will reduce aortic diastolic pressure therefore coronary flow from the hypertensive right ventricle occurring during systole through the ventriculocoronary connections may be necessary to sustain adequate myocardial perfusion. In a coronary circulation that is wholly or in part right ventricular dependent, it is the blood that gets into the right ventricle at systemic or above-systemic right ventricular systolic pressure that supplies the dependent myocardium in a retrograde fashion. Unfortunately this process can lead to further coronary arterial distortion. The management corollary to this is clear: interference of flow into the right ventricle or a reduction in right ventricular systolic pressure in situations in which the coronary circulation is dependent on the right ventricle may result in myocardial ischemia, infarction, and death.
TABLE 40.1 A Right Ventricular–Dependent Coronary Circulation | ||||
|---|---|---|---|---|
|
There is unequivocally a predisposition to ventriculocoronary connections among patients who have the smallest right ventricles. Thus, it is unlikely but not impossible to define such abnormal communications in patients with a normal-sized right ventricle or with a nearly normal right ventricle (53). It is much more likely to observe ventriculocoronary communications in patients whose ventricles have been categorized as unipartite or bipartite. Using the convention of the tricuspid Z-value, data from the CHSS demonstrated a positive correlation with ventriculocoronary connections. A negative tricuspid Z-value correlated with the presence of ventriculocoronary connections. Data from this study support the observation that the smallest tricuspid valves (i.e., the most negative Z-value) are observed in patients with the smallest right ventricles (5). Data
provided by the CHSS indicated that, of the 145 patients for whom this information was available, ventriculocoronary connections were observed in 45%. In 9% of the 145 patients, the coronary circulation was considered wholly right ventricular dependent. Ventriculocoronary connections may involute after successful right ventricular decompression (whether by pulmonary valvotomy or tricuspid valve excision or avulsion). Such involution has been demonstrated in the newborn and in the older patient. After a reduction in right ventricular pressure, there is always the possibility that flow from the coronary artery to the right ventricle might occur or become exaggerated, and this phenomenon has been recognized.
provided by the CHSS indicated that, of the 145 patients for whom this information was available, ventriculocoronary connections were observed in 45%. In 9% of the 145 patients, the coronary circulation was considered wholly right ventricular dependent. Ventriculocoronary connections may involute after successful right ventricular decompression (whether by pulmonary valvotomy or tricuspid valve excision or avulsion). Such involution has been demonstrated in the newborn and in the older patient. After a reduction in right ventricular pressure, there is always the possibility that flow from the coronary artery to the right ventricle might occur or become exaggerated, and this phenomenon has been recognized.
Perhaps of more concern is a determination of the timing of the occurrence of coronary arterial obstructive lesions, coronary artery stenosis, or interruption. Such coronary arterial obstructive lesions can occur in fetal tissues and these lesions have been identified clinically by both angiography and by histopathology in hearts from patients who die in the first few hours and days after birth. Thus, such changes should not be interpreted as a later, acquired postnatal phenomenon (49). Obviously, some changes may be acquired late, but clearly obstructive coronary arterial lesions may be present and identified in the immediate newborn.
Clinical Features
Physical Examination
Newborns with pulmonary atresia and intact ventricular septum become cyanotic and hypoxemic coincident with functional and anatomic closure of the patent arterial duct. In rare patients in whom the interatrial communication is truly restrictive, the cardiac output may be affected as well by restricting the obligatory right-to-left shunt.
There is no known sex predilection, and there is no identified genetic predisposition, although familial cases have been described as well as an occurrence in monozygotic twins (54). It has been assigned to a single gene theory in at least one instance (55). Infants typically are born at term. Cyanosis is usually apparent within hours of birth and is progressive. Dyspnea is not conspicuous without significant acidosis, reduced cardiac output, or pulmonary hypoplasia but tachypnea may be prominent.
In the absence of profound cardiac enlargement, the left precordium will not bulge. The left ventricle may be enlarged, and at the apex, its impulse may be forceful. The first and second heart sounds are single. A pansystolic murmur often is audible at the left lower sternal border, consistent with tricuspid regurgitation. In infants with severe tricuspid regurgitation, the murmur of tricuspid regurgitation is conspicuous, sometimes associated with a thrill, and a tricuspid diastolic rumble may be audible. A murmur of the arterial duct may be distinguished in the second and the third left intercostal space, especially after prostaglandin has been administered to promote ductal patency. Unless the atrial septum is profoundly restrictive, affecting cardiac output, the caliber of the arterial pulses is normal. The liver is not particularly enlarged unless there is severe tricuspid insufficiency or a restrictive foramen ovale.
The most striking and consistent finding before prostaglandin is administered is hypoxemia refractory to increased inspired oxygen concentration and a mild degree of hypocarbia, reflecting the tachypnea. Significant metabolic acidosis usually indicates progressive hypoxic cellular damage signifying imminent death in the absence of intervention.
Radiographic Features
The chest radiograph may demonstrate a heart that is only mildly enlarged or one that fills the entire chest cavity. In the former, the pulmonary vascular markings are reduced, which may be confirmed on the lateral radiograph, where hilar pulmonary arterial markings are sparse. In the latter situation, with massive cardiomegaly, it may be difficult to define enough pulmonary parenchyma to evaluate the lung markings. The differential radiographic diagnosis of extreme cardiomegaly in the newborn includes the conditions shown in Table 40.2.
TABLE 40.2 Differential Diagnosis of Massive Cardiomegaly in the Newborn | ||||||
|---|---|---|---|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


