Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is a progressive disease of the pulmonary vasculature in which an ever increasing resistance to circulatory flow imposes a mounting afterload for the right heart to overcome. Without therapy, and frequently despite it, patients with PAH suffer progressive and inexorable right heart failure, functional decline, and ultimately die. Although rapid progress has resulted in the availability of therapy that can improve the outlook for many patients, long delays in disease recognition are common, exposing patients to prolonged suffering and potentially irreversible harm.
PAH is one of several possible causes of pulmonary hypertension. Pulmonary hypertension is defined as a mean pulmonary artery pressure greater than or equal to 25 mm Hg at rest. It can be due to diseases primarily isolated to the pulmonary vasculature itself, as in PAH, or can be a complication of other diseases, including hypoxemic lung disorders (e.g., chronic obstructive pulmonary disease [COPD]), left heart disease (e.g., systolic, diastolic, or valvular dysfunctions), or thromboembolism.1,2 Identification of its cause is essential, as appropriate therapy for pulmonary hypertension is aimed at its underlying cause—be that repair of a stenotic mitral valve, bronchodilators for obstructive lung disease or, in the case of PAH, the use of advanced therapies targeted at the pulmonary vasculature.
CLASSIFICATION OF THE PULMONARY HYPERTENSIVE DISEASES
A “sclerosis of the pulmonary arteries” (“Uber Sklerose der Lungen Arterie”) without identifiable cause was first described by Ernst von Romberg in 1891.3 Exclusively descriptive reports of pathologic findings continued until the 1950s when the development of catheterization techniques allowed for hemodynamic evaluation. Using such methods, Dresdale and colleagues described a hypertensive vasculopathy of the pulmonary circulation involving vasoconstriction, elevation of pulmonary arterial pressures (PAPs), and a measurable response to the injection of the nonselective alpha adrenergic antagonist tolazoline.4 No cause could be identified for the pulmonary arteriopathy and the term primary pulmonary hypertension (PPH) was introduced.5
Subsequent classification schemes for diseases-causing pulmonary hypertension have been adopted by international consensus panels. These have evolved from systems based primarily on histopathologic findings to a current model that emphasizes the grouping of entities according to similarities in hemodynamic and clinical characteristics (Table 72-1).6 Importantly, accurate classification of pulmonary hypertension is essential to guide the rational and appropriate use of medications.
a5th WSPH Nice 2013. Main modifications to the previous Dana Point classification are in bold.
BMPR2, bone morphogenetic protein receptor type II; CAV1, caveolin 1; ENG, endogin; HIV, human immunodeficiency virus; PAH, pulmonary arterial hypertension.
Source: Adapted with permission from Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–S54.
It is important to note the specific nomenclature purposefully adopted in the current classification scheme. The group of patients with PAH is identified as distinct from those patients with other causes of pulmonary hypertension (e.g., due to chronic left heart or respiratory disease). This grouping recognizes similarities in the histologic and many clinical features of patients with identifiable genetic causes of PAH (i.e., those with heritable PAH), collagen vascular or other diseases known to be associated with PAH (Associated PAH), and patients in whom no known associated entity or genetic cause has been found. This last group is referred to as having idiopathic PAH (IPAH). IPAH replaces the previously (and often loosely) used term “PPH.” Abandonment of the term “primary” also is important as a means of discouraging the use of the confusing and clinically inappropriate term “secondary” pulmonary hypertension. Use of such “primary” and “secondary” groupings may inappropriately suggest clinical similarities among the many very different diseases previously referred to as “secondary” pulmonary hypertension (e.g., patients with COPD and those with congenital heart disease). It may also promote a failure to recognize important similarities in clinical features (including appropriate treatment) between what was previously called “primary” pulmonary hypertension and entities inappropriately labeled “secondary” (e.g., patients with Eisenmenger syndrome or HIV infection).
DETERMINANTS OF PULMONARY ARTERY PRESSURE AND PULMONARY VASCULAR RESISTANCE
The pulmonary circulation normally is a high-flow, low-resistance, low-pressure system that carries blood into the pulmonary microcirculation where the blood takes up oxygen and unloads excess carbon dioxide. From early childhood to the fifth decade of life, the mean PAP is approximately 20 mm Hg.7 PAP is the product of cardiac output (CO) and pulmonary vascular resistance (PVR) as shown in equation 1 (Eq. 1), where PVR is the vascular resistance of the entire lung, including the pulmonary arteries, capillaries, and veins.
From this equation, it is clear that PAP can be raised by an increase in CO or an increase in arterial, capillary, or venous resistance. It would be expected then, that during periods of increased CO, such as during strenuous exercise, there would be a significant increase in PAP. However, in a normal healthy individual, the PAP is only slightly increased during periods of increased CO due to the compensatory increase in the cross-sectional area of the pulmonary vascular bed (due to recruitment of previously unperfused vessels and distension of vessels) resulting in a decreased PVR. Arterial distension and optimal recruitment are dependent upon the compliance of the blood vessel walls. Loss of this compliance due to vascular remodeling leads to pulmonary hypertension. As demonstrated in Eq. 1, an increase in PVR of any of the three components of the pulmonary vasculature can lead to an increase in PAP.8
Eq. 1 demonstrates the physical laws that govern blood flow in the lungs. To further understand and use this equation, we must know its physical and anatomical foundations. When a liquid (e.g., blood) flows through a cylindrical tubular structure (e.g., a blood vessel), the resistance (e.g., PVR) is inversely proportional to the fourth power of the radius of the lumen of the tube. This is demonstrated by the Poiseuille equation (Eq. 2), where L is the length of the tube (or vessel), r is the inner radius of the tube, and η is the coefficient of viscosity of the liquid (blood). Therefore, even small changes in the radius of a vessel can significantly change the PVR.8
PULMONARY VASCULAR STRUCTURAL AND FUNCTIONAL CHANGES IN PULMONARY HYPERTENSION
Regardless of the initial genetic or pathogenic trigger, the increased PVR seen in pulmonary hypertension can be attributed to the collective effects of sustained vasoconstriction, vascular remodeling, in situ thrombosis, and increased arterial wall stiffness (Fig. 72-1).8–13 A rise in cytosolic free Ca2+ concentration ([Ca2+]cyt) in pulmonary arterial smooth muscle cells is a major trigger for vasoconstriction and a key stimulus for pulmonary arterial smooth muscle cell proliferation and migration, which contributes to vascular remodeling.
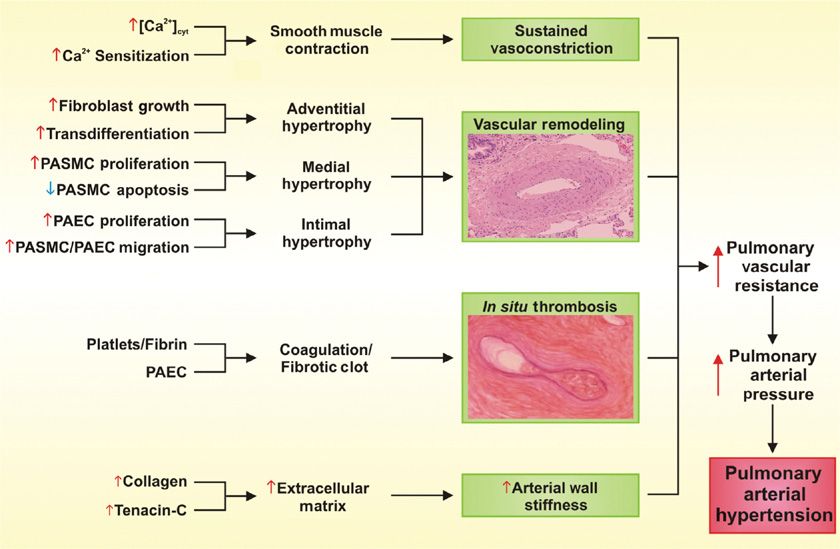
Figure 72-1 Schematic illustration of pathophysiologic components contributing to the development of increased pulmonary vascular resistance, pulmonary arterial pressure, and pulmonary arterial hypertension. (In situ thrombosis figure reproduced with permission from Zwicke D. PAH and pregnancy: Physiological changes, challenges, and outcomes. Advances in Pulmonary Hypertension. Fall; 2011;10(3).)
 SUSTAINED VASOCONSTRICTION
SUSTAINED VASOCONSTRICTION
Pulmonary vasoconstriction can be a major contributor to increased PVR and hence PAP. Vasoconstrictive lesions include medial hypertrophy involving an increase in the number and size of pulmonary arterial smooth muscle cells. The elevated PAP and sustained vasoconstriction in PAH can, in turn, enhance pulmonary arterial smooth muscle cell hypertrophy and hyperplasia.14 Marked smooth muscle hypertrophy can eventually cause medial atrophy, fibrosis, and the subsequent thinning of the media and dilation of the vessel lumen. Extension of pulmonary arterial smooth muscle cells into vessels normally only partially muscularized or nonmuscularized is a common and often prominent feature of precapillary vessels (Fig. 72-2).15
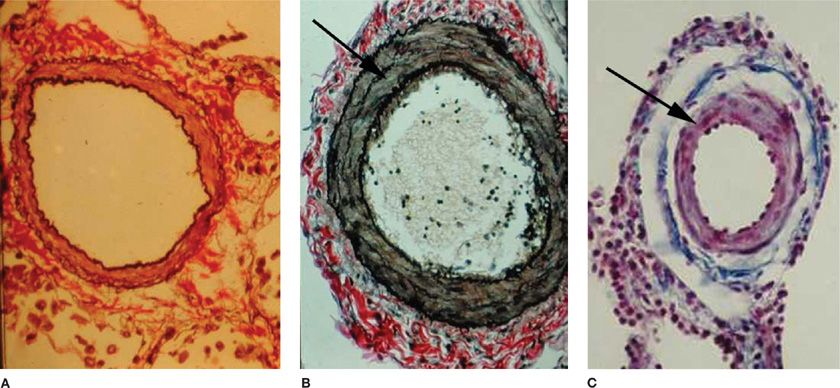
Figure 72-2 Vascular remodeling. As compared with a normal vessel (A), hypertrophy of smooth muscle cells (arrow) is seen in the pulmonary artery of a patient with pulmonary arterial hypertension (B). Extension of muscle (arrow) into normally nonmuscularized small intra-acinar pulmonary vessels is another prominent feature of pulmonary arterial hypertension (C). (Reproduced with permission from Taichman DB, Snow JL, Pietra GG. Histopathology of pulmonary arterial hypertension. In: Pulmonary Vascular Disease, Mandel J, Taichman DB. Philadelphia: WB. Saunders; 2006.)
Hypoxic pulmonary vasoconstriction is an adaptive mechanism important for redirecting blood flow away from poorly ventilated areas of the lung and into the better ventilated areas to maximize the ventilation–perfusion matching and optimize oxygenation of blood.16–19 Hypoxia induces vasoconstriction in isolated pulmonary arteries without the endothelium and also induces contraction in a single isolated pulmonary arterial smooth muscle cell model, indicating that hypoxic pulmonary vasoconstriction is an intrinsic property of pulmonary arterial smooth muscle cells. In chronic hypoxia, two factors contribute to increased PAP: vascular remodeling due to pulmonary arterial smooth muscle cell proliferation and sustained vasoconstriction and structural changes that develop within a matter of weeks.20,21 This remodeling is characterized by thickening of the media of the small pulmonary arteries and arterioles, and by peripheral extension of muscle into minute pulmonary vessels that are normally devoid of muscle (Fig. 72-2).22 Although the precise mechanism by which hypoxia induces pulmonary vasoconstriction is still somewhat unclear, the signaling pathways initiated in response to acute and chronic hypoxia seem to relate, at least in part, to disrupted Ca2+ homeostasis.8,23
 VASCULAR REMODELING
VASCULAR REMODELING
The thickness and tissue mass of the pulmonary arterial walls are maintained at an appropriate level by a fine balance between proliferation and apoptosis of fibroblasts, pulmonary arterial smooth muscle cells, and pulmonary arterial endothelial cells. Thickening of the wall and luminal narrowing and eventual obliteration can occur if the balance is tipped in favor of cell proliferation. These structural changes that lead to hypertrophy and/or luminal occlusion are referred to as pulmonary vascular remodeling.24–26 The precise cellular and molecular mechanisms contributing to vascular remodeling are extremely complex. However, vasoconstriction and cellular proliferation share a common pathway. Increased proliferation and hypertrophy of pulmonary arterial smooth muscle cells have been implicated in the development of PAH and these processes, like vasoconstriction, relate in part to disturbed Ca2+ homeostasis.7 Resting [Ca2+]cyt is increased in proliferating pulmonary artery smooth muscle cells compared to growth-arrested cells, demonstrating a role for enhanced Ca2+ in both proliferation and vasoconstriction.27,28 In addition to increased proliferation, decreased apoptosis has been implicated in the development and maintenance of severe pulmonary hypertension,29 and induction of apoptosis has been shown to promote the regression of hypertrophied pulmonary vascular walls in animal experiments.30 Hypoxia can induce proliferation of pulmonary arterial adventitial fibroblasts, and in pulmonary hypertension patients, hypoxia induces the appearance of α-smooth muscle actin in the proliferative and matrix-producing fibroblasts, suggesting these cells have transdifferentiated into myofibroblasts.31 Therefore, fibroblasts may play an important role in vascular remodeling due to hypoxia.
Complex lesions such as plexiform lesions also contribute to vascular remodeling. Plexiform lesions are aneurysmatic dilations of a muscular artery that can occur in very small arteries and arterioles.8,32 These lesions are often found in patients with IPAH, but they also occur in the lungs of patients with severe PAH associated with left-to-right cardiac shunts, HIV infection, liver cirrhosis, and scleroderma. Although not pathognomonic, the plexiform lesion has been the focus of many studies of the cellular and molecular pathogenesis of PAH.33–36 They contain collections of proliferating endothelial and smooth muscle cells, together with myofibroblasts and matrix proteins that can partially or completely occlude the vessel lumen. Narrowing or complete obliteration of the parent vessel by intimal thickening is a frequent associated finding, as is destruction of its media. Plexiform lesions often coexist with other obliterative vascular changes such as concentric laminar intimal thickening.37
The origin of plexiform lesions is complex and somewhat controversial. Originally, plexiform lesions were considered a congenital malformation.38 Currently, there are investigators who believe that plexiform lesions develop due to the proliferation of pulmonary arterial smooth muscle cells that transform into myofibroblasts.39–41 Other investigators propose that, in IPAH patients, plexiform lesions develop due to an endothelial-initiated response to cytokines, growth factors, or vascular injury.39,41,42 Endothelial cells isolated from the plexiform lesions of IPAH patients proliferate in a monoclonal fashion. However, plexiform lesions from patients with forms of pulmonary hypertension other than IPAH develop from a polyclonal cell population, suggesting different mechanisms contribute to plexiform lesion development in different forms of PAH.8,43
 IN SITU THROMBOSIS
IN SITU THROMBOSIS
Monoclonal proliferation of pulmonary arterial endothelial cells, pulmonary arterial smooth muscle cell migration, and accumulation of circulating inflammatory cells, platelets, and progenitor cells can result in occlusion of smaller vessels.11,42 Thrombosis is frequently seen within the small vessels of patients with PAH. This occurs without evidence of a remote (embolic) source of the thrombus,44–47 suggesting a local imbalance of pro- and anticoagulant forces. Activation and altered function of the endothelium leading to a shift from anti- to procoagulant activities might be due to the effects of shear stress associated with elevated pressure and/or flow. In addition to altered endothelial cell activity, platelets also promote thrombus formation by releasing vasoactive and mitogenic factors such as thromboxane metabolites and serotonin.48 These, as well as other platelet-derived products (e.g., platelet-derived growth factor [PGDF], transforming growth factor-beta (TGFβ), and vascular endothelial growth factor [VEGF]) likely also contribute to the remodeling of vessel walls seen in PAH.
 INCREASED ARTERIAL WALL STIFFNESS
INCREASED ARTERIAL WALL STIFFNESS
Increased arterial wall stiffness can also contribute to increased PVR.49,50 The normal turnover of extracellular matrix proteins is accelerated with remodeling of the vasculature in PAH patients.51,52 The expression of tenascin-C, for example, is increased in experimental pulmonary hypertension induced by either monocrotaline in rats or increased blood flow in swine.53–55 Indeed, inhibition of tenascin-C expression by antisense RNA ameliorates monocrotaline-induced pulmonary vascular lesions.56 Elevated levels of this extracellular matrix protein are also seen on pulmonary arteries of patients with PAH.57,58 Decreased vascular compliance resulting in the inability to recruit previously unperfused vessels due to increased parenchymal stiffness can also contribute to the development of PAH.11,42 As a result, there is little capacity for distention and modest increases in pulmonary blood flow can elicit disproportionate elevations in PAPs. This situation is in marked contrast to that of the normal pulmonary circulation, in which an amputation of considerable lung parenchyma rarely suffices, per se, to raise PAPs to pulmonary hypertensive levels, as the remaining vessels retain their normal high capacitance. In some interstitial lung diseases, such as progressive systemic sclerosis, the parenchymal disease and the pulmonary vascular disease can evolve independently. In other connective tissue disorders, such as systemic lupus erythematosis, combinations of interstitial disease and intrinsic vascular abnormalities can contribute to pulmonary hypertension. While there are many structural changes in the pulmonary vasculature that lead to the development of PAH, vasoconstriction and vascular remodeling characterized by intimal, medial, and adventitial hypertrophy are the major structural changes that contribute to the increased PVR/PAP seen in PAH patients.
No single histologic feature distinguishes between the clinical PAH diagnoses. Each of the changes described can be seen in varying proportions in all clinical forms of PAH. For example, the vascular remodeling seen in chronic hypoxia-induced pulmonary hypertension is virtually indistinguishable from that seen in IPAH.8 The predominant causes of the increased PVR in patients with PAH are sustained vasoconstriction, vascular remodeling, in situ thrombosis, and increased arterial wall stiffness. Persistent pulmonary hypertension, regardless of the cause, can ultimately lead to the development of cor pulmonale with hypertrophy of the right ventricle, its eventual dilation, and ultimately its failure (Fig. 72-3).
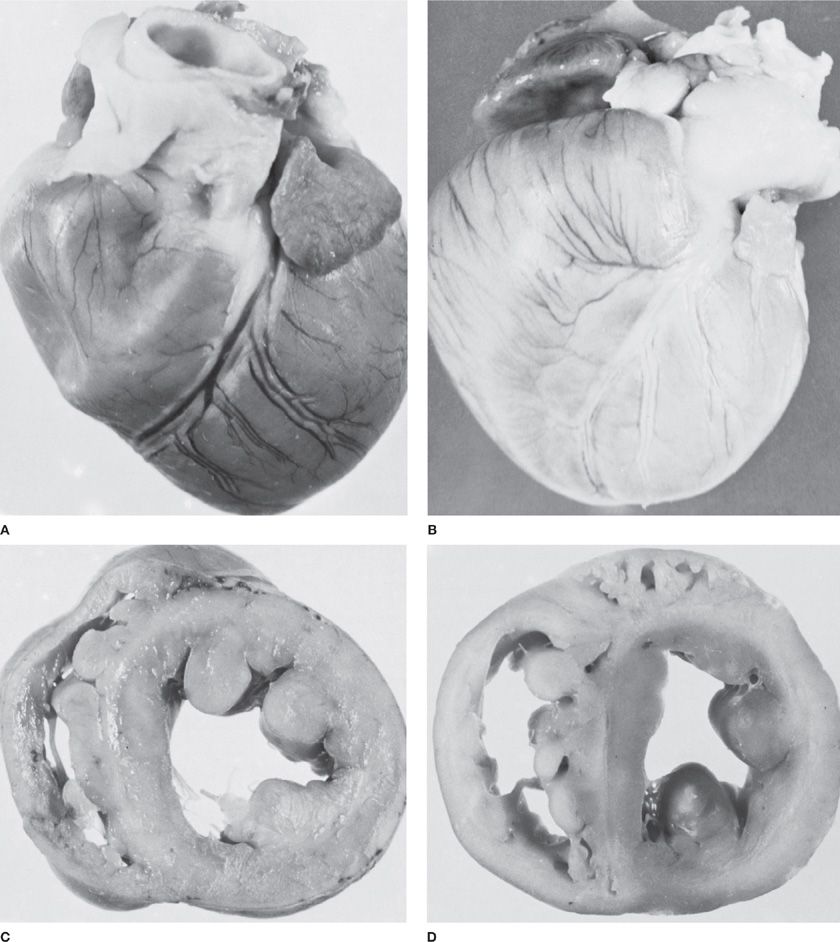
Figure 72-3 Cor pulmonale in experimental pulmonary arterial hypertension in the dog. A. Normal heart. B. Chronic cor pulmonale secondary to severe pulmonary arterial hypertension. C. Cross section of normal heart to show thin wall of the right ventricular cavity. D. Cross section of heart with chronic cor pulmonale to show hypertrophy of the right ventricular myocardium and enlargement of the right ventricular cavity. (Used with permission of Dr. B. Atkinson.)
PATHOGENIC MECHANISMS OF PULMONARY HYPERTENSION
The pathogenic mechanisms leading to pulmonary hypertension have been conceptualized in six categories (Table 72-2): (1) passive, due to obstruction to pulmonary venous outflow (e.g., fibrosing mediastinitis, mitral stenosis, or left heart failure); (2) hyperkinetic, due to abnormally high pulmonary blood flow (e.g., left-to-right shunts); (3) obstructive, due to pulmonary thromboembolic disease; (4) obliterative, due to destruction of the pulmonary vascular bed by parenchymal proliferative disease; (5) vasoconstrictive, due to hypoxic vasoconstriction; and (6) idiopathic (i.e., without discernible cause).59 Each mechanism leads to pulmonary vascular remodeling discussed previously, though the progression and reversibility may vary between entities. Other anatomic alterations can be seen in several categories and, over time, distinctions between categories tend to become blurred (e.g., thrombosis may complicate obliterative vascular disease). Also, by the time pulmonary hypertension becomes manifest clinically, the pulmonary arterial tree has undergone considerable remodeling that limits its area and distensibility.
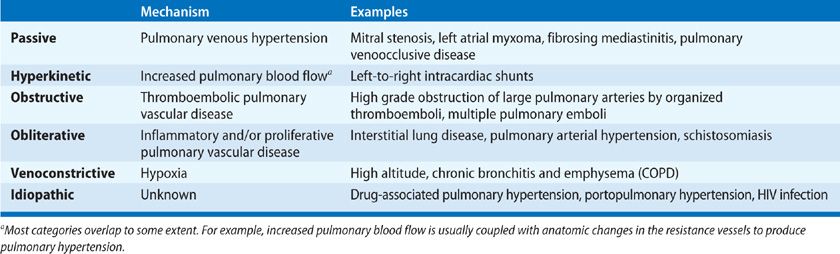
These mechanisms share commonality with the clinical classification of pulmonary hypertension, though overlap of mechanisms occur. The most clearly linked is chronic thromboembolic pulmonary hypertension (CTEPH) with the obstructive mechanism. The most common form of pulmonary hypertension, that related to left heart failure, can also closely be tied to its passive mechanism along with fibrosing mediastinitis, which is classified in group 5 with unclear multifactorial mechanisms. Pulmonary hypertension owing to lung diseases and/or hypoxia, group 3, can be understood as having two primary pathogenic mechanisms, vasoconstrictive in the chronic hypoxic subset and obliterative in the parenchymal disease/fibrosis subset, though these two often overlap. Group 5, pulmonary hypertension with unclear multifactorial mechanisms by definition contains multiple pathogenic mechanisms. Group 1 remains the most diverse and difficult to equate to a particular mechanism as hyperkinetic, vasoconstrictive, obliterative, and other idiopathic mechanisms all contribute to disease pathophysiology.
GENETIC, CELLULAR, AND MOLECULAR MECHANISMS OF PULMONARY ARTERIAL HYPERTENSION
The development of PAH involves increased PVR due to sustained vasoconstriction, vascular remodeling, in situ thrombosis, and increased atrial wall stiffness (Fig. 72-1). Abnormalities in the expression of numerous vasoactive mediators, vasoconstriction mediators, growth factors, and cytokines cause, or result from changes in pulmonary arterial endothelial cells, pulmonary arterial smooth muscle cells, and platelet function and together result in a thickened vessel wall and markedly narrowed or even completely obliterated lumen. Some of the mechanisms for this combination of uncontrolled vasoconstriction, cell proliferation, and thrombosis are highlighted here.
 IMBALANCE OF VASOACTIVE MEDIATORS
IMBALANCE OF VASOACTIVE MEDIATORS
Relative deficiencies of factors with vasodilatory properties and a simultaneous excess in those promoting vasoconstriction have been noted in both animal models and patients with PAH. In addition to vasoconstriction/dilation, these same factors influence cell proliferation and thrombosis. Deficiencies in the production of the potent vasodilators nitric oxide (NO) and prostacyclin have each been observed, and indeed both substances have been used as a basis for effective therapies for PAH. Normally produced by vascular endothelial cells, each promotes the formation of cyclic nucleotides (cGMP and cAMP) by pulmonary arterial smooth muscle cells resulting in their relaxation and vasodilation. In addition, both prostacyclin and NO inhibit pulmonary arterial smooth muscle cell proliferation and platelet aggregation. In patients with PAH, the chronic administration of prostacyclin analogs improves hemodynamics, exercise capacity, and survival. The overexpression of NO synthase (NOS) by transgenic animals protects against the development of hypoxia-induced pulmonary hypertension, whereas mice lacking the gene for this enzyme develop severe pulmonary hypertension upon exposure to mild hypoxia.60–62 In rats, monocrotaline-induced pulmonary hypertension can be prevented, or even reversed, with the administration of endothelial progenitor cells overexpressing human endothelial NOS (eNOS).63 Inhaled NO has been proposed for the treatment of PAH and persistent pulmonary hypertension of the newborn,64 however, inhaled NO has limitations regarding dose and duration of the exposure.65 The inorganic anion nitrite (NO2–) is an oxidative product of NO metabolism and can function as an intravascular reservoir of NO bioactivity that can be converted to NO under physiologic and pathologic conditions.65 Studies have shown that inhaled nebulized nitrite can prevent and reverse established pulmonary hypertension in the monocrotaline-induced rat pulmonary hypertension model.66,67
Vasoactive intestinal protein (VIP) also promotes vasodilation and inhibits smooth muscle proliferation and platelet aggregation. VIP levels are reduced in patients with PAH. In a preliminary study of 8 patients with IPAH, treatment with inhaled VIP improved hemodynamics and exercise capacity.68 The acute effect of inhaled VIP was confirmed in a subsequent study that included patients with multiple etiologies of pulmonary hypertension including IPAH, congenital heart disease, CTEPH, and pulmonary hypertension related to parenchymal lung disease, although the modest effect on hemodynamics had questionable clinical relevance.69
 INCREASE IN VASOCONSTRICTION MEDIATORS
INCREASE IN VASOCONSTRICTION MEDIATORS
In addition to deficiencies in vasodilators, excesses of other mediators capable of promoting vasoconstriction, smooth muscle proliferation, or platelet aggregation have been noted in patients with PAH. Thromboxane, an arachidonic acid metabolite produced by endothelial cells and platelets, causes vasoconstriction, platelet aggregation, and is a smooth muscle mitogen. Increased thromboxane metabolites have been documented in the urine of patients with PAH.48 Attention was focused on the effects of serotonin (5-HT) on the pulmonary circulation by the epidemic of PAH in patients who ingested the appetite suppressants aminorex and fenfluramine.70 These agents increase plasma 5-HT levels by inducing the release of 5-HT from platelets and interfering with its reuptake.71 The 5-HT hypothesis was supported by the occurrence of pulmonary hypertension in fawn-hooded rats with an inherited defect in the platelet storage of 5-HT, and by an increase in circulating 5-HT in a patient with platelet storage disease and PAH.72 5-HT causes vasoconstriction and is a smooth muscle mitogen (Fig. 72-4). A key regulator of 5-HT action is the 5-HT transporter (5-HTT) whose expression is increased above normal in platelets and the pulmonary arteries of patients with IPAH. The overexpression of the 5-HTT gene in recombinant mice results in worsened hypoxia-induced pulmonary hypertension73 whereas loss of the gene’s function is protective against hypoxia or monocrotaline-induced disease.74,75 A polymorphism in the 5-HTT gene that increases its activity may confer an increased susceptibility to the development of pulmonary hypertension in patients with COPD.76 While some studies have suggested a similar role in IPAH,77 larger data sets have not found such an association.78 Blockade of the 5-HT receptor has successfully prevented the development and progression of experimental pulmonary hypertension in rats, yet a human trial was not able to show significant decreases in PVR.79,80
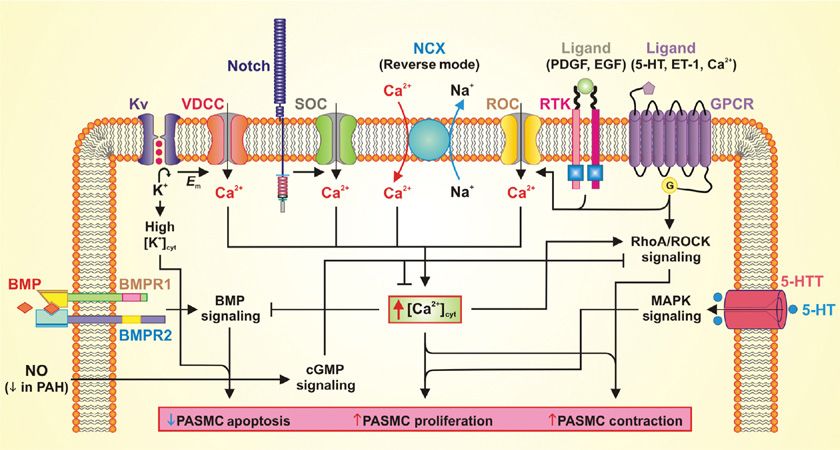
Figure 72-4 Potential mechanisms involved in the development of pulmonary arterial hypertension. Schematic diagram depicting potential mechanisms involved in the development of pulmonary arterial hypertension. BMP, bone morphogenic protein; BMPR, bone morphogenic protein receptor; cGMP, cyclic guanosine monophosphate; Em, membrane potential; ET-1, endothelin 1; EGF, epidermal growth factor; GPCR, G-protein–coupled receptor; 5-HT, hydroxytryptamine (serotonin); 5-HTT, hydroxytryptamine (serotonin) transporter; Kv, voltage-gated K+ channel; MAPK, mitogen-activated protein kinase; NO, nitric oxide; NCX, Na+/Ca2+ exchanger; PAH, pulmonary arterial hypertension; PASMC, pulmonary arterial smooth muscle cell; PDGF, platelet-derived growth factor; ROC, receptor-operated Ca2+ channel; RTK, receptor tyrosine kinase; SOC, store-operated Ca2+ channel; VDCC, voltage-dependent Ca2+ channel.
Endothelin-1 (ET-1) is one of the most potent endogenous vasoconstrictors known. Its expression is increased in the blood and tissues of patients with idiopathic and other forms of PAH and correlates with disease severity.81–83 In addition to its vasoconstricting properties, ET-1 is mitogenic for both pulmonary arterial smooth muscle cells and fibroblasts (Fig. 72-4).84,85 ET-1 administration or overexpression in animal models has been shown to result in fibrosis, inflammation, and platelet aggregation.86,87 ET-1 binds endothelin A (ETA) and B (ETB) receptors on the surface of pulmonary arterial smooth muscle cells, resulting in potent vasoconstriction. ETB receptors on pulmonary arterial endothelial cells increase the production of nitric oxide, resulting in vasodilatation. ETB receptors are also active in the clearance of endothelin. The net effect of endothelin’s vasoconstricting or dilating actions may be both site and context dependent, as both the distribution and relative expression of the ETA and ETB receptors differ according to vessel location in normal lung tissue, and are altered in patients with IPAH.88,89 Indeed, both selective ETA and dual ETA/ETB inhibition improve the hemodynamic derangements and clinical outcome of patients with PAH.90–92
 INCREASED EXPRESSION OF GROWTH FACTORS
INCREASED EXPRESSION OF GROWTH FACTORS
Growth factors that promote the maturation and stabilization of the developing vasculature have also been implicated in the pathogenesis of PAH. Elevations in angiopoietin 1 and its ligand TIE2 correlate with disease severity in patients with multiple forms of PAH.93 Angiopoietin 1 is overexpressed in most forms of nonfamilial PAH,93,94 however, how angiopoietin becomes elevated in these patients is not clear. Several lines of evidence suggest that angiopoietin 1 regulates pulmonary arterial smooth muscle cell hyperplasia in PAH.9 Interestingly, in an animal model of PAH induced by monocrotaline, the overexpression of angiopoietin is actually protective.95 Whether this discrepancy represents differences in human versus animal tissues or the differing insults to the vasculature involved is not clear. Another modulator of development, VEGF and its target, tyrosine kinase receptor, are increased in the pulmonary vasculature of patients with PAH. Increased VEGF expression has been reported specifically within plexiform lesions34,96 where its proangiogenic properties are hypothesized to mediate disordered endothelial cell proliferation.97 Whether such changes are primary, secondary, or indeed detrimental is not entirely clear. Like elevations in angiopoietin, increases in the expression of VEGF thought to be deleterious in some situations might be beneficial in others that promote the development of pulmonary hypertension. In animal models of hypoxia, the inhibition of VEGF signaling results in proliferative vascular abnormalities98 and promotion of VEGF signaling is protective against the development of monocrotaline-induced PH.99
PDGF along with its receptor, tyrosine kinase, are also found to be increased in the pulmonary vasculature of patients with PAH (Fig. 72-4).100 PDGF has been implicated in smooth muscle cell proliferation and migration, which is thought to contribute to PAH pathogenesis. The tyrosine kinase inhibitor imatinib has a particular affinity for the PDGF receptor and several small studies have identified a potential benefit in PAH patients.101 Early human trials have also shown improvements in hemodynamic measurements after treatment.102
Cellular microparticles recently have been implicated as vasoactive mediators in PAH. Microparticles represent vesicle fragments of the cell membrane containing various proteins and antigens that are involved in cellular communication.103 These small particles are released during cell activation or apoptosis and can be derived from multiple cell lines including endothelial cells, platelets, leukocytes, red blood cells, and fibroblasts. Circulating levels of endothelial microparticles are increased in PAH, which may represent endothelial dysfunction and has also been correlated with disease survival.104 Microparticles, when isolated from rats with hypoxia-induced pulmonary hypertension, have been shown to impair endothelium-dependent vasorelaxation in pulmonary arteries and decrease NO production.105 Circulating microparticles have also been linked to inflammatory signaling in the lung.106
 INCREASED CYTOKINES AND INFLAMMATION
INCREASED CYTOKINES AND INFLAMMATION
The close association of systemic inflammatory disorders to IPAH has implicated inflammation as an important factor in vascular remodeling. Numerous cytokines have been implicated in the pathogenesis of PAH including tumor necrosis factor alpha, and interleukins 1b, 2, 4, 6, 8, 10, and 12p70. Early data showed increased levels of IL-1β and IL-6 in IPAH patients107 and higher IL-6 levels were later found to be associated with mortality.108 The chemokine fractalkine (CX3CL1) is elevated in CD4 and CD8 T-cells of PAH patients and further studies have shown that CX3CL1 may promote proliferation in pulmonary arterial smooth muscle cells.109,110 Monocyte chemotactant protein (MCP)-1 is also elevated in serum and lung samples from IPAH patients and may influence monocyte and T-cell recruitment to the diseased lung.111 Increased expression of Regulated upon Activation, Normal T cell Expressed and Secreted (RANTES) has been found in lung samples from PAH patients.112 As a chemoattractant for monocytes and T-cells, RANTES is thought to promote inflammation, but may also play a role in the synthesis of endothelin-1, a potent vasoconstrictor.
FUNCTIONAL CHANGES IN MEMBRANE RECEPTORS AND ION CHANNELS
Important functional changes in membrane receptors and ion channels in IPAH are discussed below.
 Ca2+ CHANNELS IN IPAH
Ca2+ CHANNELS IN IPAH
Sustained vasoconstriction and vascular remodeling are both directly mediated by pulmonary arterial smooth muscle cell contraction and proliferation. A rise in [Ca2+]cyt due to Ca2+ influx through various Ca2+-permeable channels in the plasma membrane is a major trigger for pulmonary arterial smooth muscle cell contraction and a key stimulus for pulmonary arterial smooth muscle cell proliferation.113 Therefore, understanding how the regulation of Ca2+ homeostasis is disrupted in IPAH is important to understanding how IPAH develops. Pulmonary arterial smooth muscle cells isolated from IPAH patients have an elevated [Ca2+]cyt.114–117 Ca2+ influx through the plasma membrane in pulmonary arterial smooth muscle cells involves multiple Ca2+-permeable channels including (a) voltage-dependent Ca2+ channels (VDCC) regulated by changes in membrane potential,118 (b) receptor-operated Ca2+ channels (ROC) activated by interaction of agonist with membrane receptors, and (c) store-operated Ca2+ channels (SOC) activated by depletion of Ca2+ from the intracellular stores (Fig. 72-4).8 Decreased expression and/or function of K+ channels leads to sustained membrane depolarization, activation of VDCC, and increased [Ca2+]cyt. Ca2+ influx through ROC, termed receptor-operated Ca2+ entry (ROCE), and through SOC, referred to as store-operated Ca2+ entry (SOCE) is enhanced in IPAH-associated pulmonary arterial smooth muscle cells compared to those from normal controls.114–117 Several experimental studies have demonstrated increased expression of several proteins involved in ROCE/SOCE such as, TRPC3, TRPC6, STIM2, and Orai2, in pulmonary arterial smooth muscle cells from IPAH patients.114–117,119,120 In addition, a single-nucleotide polymorphism (SNP) has been identified in IPAH patients that results in enhanced expression and function of TRPC6.115
 K+CHANNELS IN IPAH
K+CHANNELS IN IPAH
Vascular tone might also be altered in IPAH by changes in the expression of voltage-gated K+ (Kv) channels as well as other types of K+ channels (Fig. 72-4). Their activation normally allows an efflux of K+ and resultant changes in intracellular Ca2+ that promote vasodilation.121 Gene expression of Kv channel family members is downregulated by hypoxia-induced pulmonary hypertension in rats,122,123 whereas induction of their expression can reverse the hemodynamic effects of hypoxia.122,124 The expression of specific Kv channels is also decreased in the lungs of patients with IPAH125–127 possibly contributing to heightened vasoconstriction resulting from a more depolarized membrane leading to increased [Ca2+]cyt due to activation of VDCC. Kv channels may also mediate the effects of certain drugs. The anorexigens dexfenfluramine and aminorex inhibit smooth muscle Kv channel activity, thus causing pulmonary vasoconstriction.128 In contrast, enhanced activity of Kv channels may be one mechanism by which sildenafil promotes vasodilation in addition to its activity as an inhibitor of phosphodiesterase-5.122 One final way in which decreased activity of Kv channels might promote the development of PAH is by inhibiting apoptosis, thus enabling unchecked pulmonary arterial smooth muscle cell proliferation. Apoptosis requires a loss in cell volume as well as the function of specific caspases, both of which require appropriate K+ movement via Kv channels.129,130
 G-PROTEIN–COUPLED RECEPTORS IN IPAH
G-PROTEIN–COUPLED RECEPTORS IN IPAH
The Ca2+-sensing receptor is a G-protein–coupled receptor in the plasma membrane that can be activated by extracellular Ca2+ (and Mg2+), polyamines (e.g., spermine), amino acids and neomycin.131–134 Activation of the Ca2+-sensing receptor results in activation of intracellular Ca2+ signaling pathways that lead to pulmonary arterial smooth muscle cell contraction, proliferation, and migration (Fig. 72-4).135,136 Like some G-protein–coupled receptors coupled to Gq (e.g., endothelin receptors), Ca2+-sensing receptor activation increases the synthesis of inositol 1,4,5 triphosphate (IP3) and diacylglycerol via phospholipase C. IP3 binds to the IP3 receptor on the sarcoplasmic reticulum (SR) membrane and releases Ca2+ from the SR to the cytosol. Depletion of Ca2+ from the SR induces Ca2+ entry via SOCE, and diacylglycerol directly activates ROCE. In addition to increasing [Ca2+]cyt via ROCE and SOCE, the extracellular Ca2+-induced activation of CaSR also activates other signal transduction pathways (e.g., Akt/mTOR and MAPK/ERK) to induce cellular proliferation.137–139 A recent study135 indicates that the extracellular Ca2+-induced increase in [Ca2+]cyt by activation of the Ca2+-sensing receptor is enhanced and the Ca2+-sensing receptor protein expression is upregulated in IPAH-pulmonary arterial smooth muscle cells compared to normal pulmonary arterial smooth muscle cells. These observations suggest that upregulated expression and enhanced function of Ca2+-sensing receptor in pulmonary arterial smooth muscle cells are involved in the development of sustained pulmonary vasoconstriction and pulmonary vascular remodeling in patients with IPAH and animals with experimental pulmonary hypertension. In addition to Ca2+-sensing receptor, many other G-protein–coupled receptors are reported to be involved in PAH such as endothelin receptor, prostacyclin receptor, and serotonin receptor.48,140,141
 Na+/Ca2+ EXCHANGER IN IPAH
Na+/Ca2+ EXCHANGER IN IPAH
The Na+/Ca2+ exchanger (NCX) is a ubiquitously expressed protein that transports Ca2+ across the plasma membrane along the electrochemical gradient of Na+ and Ca2+.142 The NCX can operate in either forward mode, transporting 3 Na+ ions into the cell and 1 Ca2+ ion out of the cell, or the reverse mode, transporting 1 Ca2+ ion into the cell and 3 Na+ ions out of the cell, based on the Na+ and Ca2+ concentration gradients and membrane potential. Due to the reverse mode of the NCX, a small increase in the cytosolic Na+ concentration ([Na+]cyt) can greatly increase [Ca2+]cyt. The canonical transient receptor potential (TRPC) channels, such as TRPC6, which is overexpressed in IPAH patients, are permeable to both Na+ and Ca2+, and for many TRPC channels, the permeability to Na+ is greater than the permeability to Ca2+. The reverse mode of the NCX has been shown to couple to TRPC6 and localized increases in [Na+]cyt result in transport of Ca2+ into the cell resulting in increased [Ca2+]cyt in pulmonary arterial smooth muscle cells.143 The NCX is overexpressed in PASMC isolated from patients with IPAH, and is found localized in caveolae along with other G-protein–coupled receptors, ROC, and SOC resulting in a functional coupling of these receptors and contributing to increased [Ca2+]cyt, resulting in increased proliferation and contraction of PASCM in patients with IPAH.116,144
ALTERED SIGNALING PATHWAYS IN IPAH
Alterations in signaling pathways have been observed in IPAH and are discussed below.
 CYCLIC GUANOSINE MONOPHOSPHATE IN IPAH
CYCLIC GUANOSINE MONOPHOSPHATE IN IPAH
IPAH is associated with abnormally low levels of the potent pulmonary arterial smooth muscle cell relaxant NO. In pulmonary arterial endothelial cells, NO produced by eNOS freely diffuses across the cell membrane to pulmonary arterial smooth muscle cells and activates intracellular soluble guanylate cyclase (sGC), which catalyzes the conversion of guanosine 5′-triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). cGMP then activates cGMP-dependent kinases, which can reduce [Ca2+]cyt, inhibit Rho signaling, and inhibit phosphorylation of myosin-binding protein, all of which lead to smooth muscle relaxation.145,146 In the lung, metabolism of cGMP is controlled by phosphodiesterase 5 (PDE-5). PDE-5 inhibitors block the breakdown of cGMP, leading to vasodilation due to an accumulation of cGMP in the tissue.147 In 2005, following several clinical studies, sildenafil was approved for the treatment of PAH.148 Tadalafil was approved for the treatment of PAH in 2009.149 Both sildenafil and tadalafil share structural similarities with cGMP and inhibit PDE-5 by competitively binding to the catalytic site.147
 PDGF AND Akt/mTOR SIGNALING IN IPAH
PDGF AND Akt/mTOR SIGNALING IN IPAH
PDGF and its receptor PDGFR have been implicated in the pathogenesis of IPAH. Previous studies have indicated that PDGF increases SOCE in rat and human pulmonary arterial smooth muscle cells, thereby promoting cell proliferation (Fig. 72-4).119,150 Binding of PDGF to PDGFR leads to activation of phosphatidylinositol 3-kinase (PI3K),151 which activates Akt. Akt promotes cell growth through the downstream mediator mammalian target of rapamycin (mTOR) or by directly phosphorylating proteins involved in cell cycle regulation and apoptosis.152 The Akt/mTOR pathway has been shown to be important for pulmonary arterial smooth muscle cell proliferation150,153 and the development of PAH.154 Inhibition of mTOR and Akt by rapamycin and Akt inhibitor VIII, respectively, abrogated the PDGF-induced increased SOCE and Ca2+ channel expression in normal and PAH-pulmonary arterial smooth muscle cells,150,155 suggesting that the PDGF-mediated increase in SOCE occurs via the Akt/mTOR pathway. Clinical trials have shown some benefit to the use of imatinib, an orally active PDGFR inhibitor, in patients with severe PAH.102
 RhoA/ROCK SIGNALING IN IPAH
RhoA/ROCK SIGNALING IN IPAH
The RhoA/ROCK signaling pathway is a major mediator of Ca2+-sensitization/desensitization and a key regulator of vascular tone (Fig. 72-4).156,157 RhoA is a GTP-binding protein that is activated following activation of G-protein–coupled receptors by vasodilators. In addition, RhoA can be activated by serotonin, which enters the cell through its transporter 5-HTT, and by an increase in [Ca2+]cyt.158 Activation of RhoA leads to activation of Rho kinase (ROCK), which increases the Ca2+ sensitivity of contraction in pulmonary arterial smooth muscle cells by inhibiting myosin light chain phosphatase and increasing the phosphorylation of myosin light chain kinase, leading to increased contraction of pulmonary arterial smooth muscle cells.20,21 Recent studies have indicated that the RhoA/ROCK signaling pathway is enhanced in patients with IPAH.159,160 Small clinical studies have demonstrated that inhibition of the RhoA/ROCK signaling pathway with fasudil, a selective ROCK inhibitor, results in acutely improved pulmonary hemodynamic variables in patients with IPAH.161,162
 NOTCH SIGNALING IN IPAH
NOTCH SIGNALING IN IPAH
Notch signaling is involved in vascular development and has been implicated in the development of IPAH (Fig. 72-4).163,164 Both Notch receptors (Notch1-Notch4) and their ligands, Jagged (Jag1 and Jag2) and Delta-like (Dll1, Dll3, and Dll4), are single-transmembrane–spanning proteins that restrict Notch signaling to adjacent cells. After ligand binding, Notch undergoes a series of proteolytic cleavages resulting in the release of the Notch intracellular domain, which translocates to the nucleus where it interacts with recombination signal binding protein for immunoglobulin kappa j region (RBPjκ) to function as a transcription factor resulting in activation of transcription of Notch target genes.163,165–167 The most commonly induced Notch target genes are the basic helix–loop–helix transcriptional repressors of the Hes (Hairy/Enhancers of Split) and Hrt (Hes-related transcription factor) gene families. In addition, Notch signaling has been shown to upregulate expression of PDGFR-β,168 which is known to be upregulated in IPAH.155,169 Notch3 signaling has recently been implicated in PAH. Lung tissue from IPAH patients displays increased Notch3 and Notch3 intracellular domain (N3ICD) expression when compared to normotensive patients.164 In addition, Notch3 and N3ICD expression is increased in two animal models of pulmonary hypertension, hypoxic-induced pulmonary hypertension in mice and monocrotaline-induced pulmonary hypertension in rats.164 This suggests that the Notch signaling pathway may be an important pathway to target for the development of new drugs for the treatment of IPAH.
GENETIC ALTERATIONS RELATED TO IDIOPATHIC AND FAMILIAL PAH
Identification of bone morphogenetic protein receptor II (BMPR2) gene mutations has led to increasing interest in genetic susceptibility to pulmonary hypertension. As a receptor for the TGFβ super family, BMPR2 is involved in diverse cell growth and differentiation processes in multiple systems. Engagement of a BMP receptor with its ligand normally results in the activation of intracellular mediators (Smads) and their translocation to the cell nucleus and regulation of the transcription of target genes. The resulting activation of some genes and the inhibition of others varies according to the BMP pathway and tissue involved. BMP signaling is essential to both normal vascular development and the maintenance of the normal adult pulmonary vasculature, likely by regulating the growth and apoptosis of endothelial and smooth muscle cells (Fig. 72-4). Loss of such regulation gives rise to pulmonary hypertension.170 Normally found primarily on endothelium and to a lesser extent smooth muscle cells, the expression of BMPR2 is reduced and its function abnormal in patients with various types of PAH, most notably those with mutations of the BMPR2 gene.171 The ability of BMP to inhibit smooth muscle proliferation and induce apoptosis is suppressed in cells isolated specifically from smaller pulmonary vessels in patients with IPAH (e.g., 1 to 2 mm, where occlusive vascular pathologic changes predominate).29,172,173 Germline mutations in BMPR2 have been identified in up to 70% of patients with familial PAH, and in 20% of patients with IPAH,174–180 as well as disease associated with anorexigens,181 congenital heart disease,182 and pulmonary venoocclusive disease (PVOD).183 Nearly 300 different mutations of BMPR2 have been identified.177
Further work has identified other genetic alterations in the TGFβ pathway including activin receptor kinase-like 1 (ALK1), endoglin (Eng), and Smad 8.184 ALK1 is a receptor for TGFβ and endoglin is its coreceptor; while Smad 8 is a downstream second messenger of BMPR2. Mutations in ALK1 and Eng have been shown to confer susceptibility to the development of PAH in patients with hereditary hemorrhagic telangiectasia.185,186 In addition, concomitant Thrombospondin-1 gene mutation (THBS1) has been detected in a small subset of familial PAH patients with BMPR2 mutations; this mutation has been proposed to further promote pulmonary hypertension development and increase genetic penetrance.187
CLINICAL EVALUATION OF PULMONARY HYPERTENSION
The symptoms and signs of PAH are nonspecific, overlapping with many more common entities. As a result, patients experience problematic delays between the time of symptom onset and appropriate diagnosis.188,189 The problem is usually first suggested by findings suggestive of pulmonary hypertension on an echocardiogram. The evaluation of PAH is predominantly, therefore, the evaluation of pulmonary hypertension more generically: a step-wise process to establishing whether pulmonary hypertension is likely present, and if so confirmation and careful evaluation of the specific cause and classification so as to guide rational therapy.
 PATIENT HISTORY IN PULMONARY HYPERTENSION
PATIENT HISTORY IN PULMONARY HYPERTENSION
Except for mild breathlessness – often attributed to being “out of shape” – PAH is generally asymptomatic until severe. Because of the nonspecific nature of the symptoms (Table 72-3), underrecognition of the disease by healthcare providers, and its confusion with other conditions, a significant delay between the onset of symptoms and the diagnosis of PAH is common. The mean time from symptom onset until PAH diagnosis was over 2 years in a National Institutes of Health (NIH) registry assembled in the 1980s.190 Twenty years later, registries in China, France, Germany, and the United States each found that similarly disappointing delays of over 2 years persist.189,191–193 As a result, diagnosis is delayed until most patients have severe functional impairment, with more than one-half reporting World Health Organization (WHO) functional class III or IV symptoms (Table 72-4).
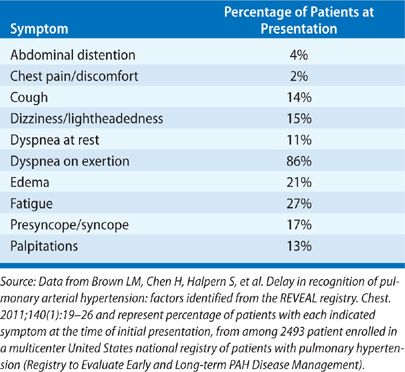
TABLE 72-4 World Health Organization Functional Classification of Patients with Pulmonary Hypertension
Source: Adapted with permission from Rich S. Primary Pulmonary hypertension: executive summary. Evian, France. World Health Organization, 1998.
Symptoms due to pulmonary hypertension are generally difficult to dissociate from symptoms of underlying pulmonary or cardiac disease. In IPAH, the first symptoms generally occur during exertion, usually as dyspnea and, less often, chest pain, dizziness, or syncope.194 Dyspnea on exertion is by far the most common presenting complaint. Often, because of the lack of other signs or symptoms, it is attributed to physical deconditioning or anxiety. Other initial complaints, particularly easy fatigability and chest discomfort, are often similarly dismissed. Angina-like or nonspecific chest pain is common in patients with severe pulmonary hypertension and generally attributed to right ventricular overload and myocardial ischemia. Chest pain might also occur due to the extrinsic compression of the left main coronary artery by an enlarged pulmonary artery.195
In time, in the absence of amelioration of the pulmonary hypertension, right-sided heart failure evolves. Syncope, or light-headedness on exertion, are less common but more ominous complications of pulmonary hypertension. They occur in patients with severe pulmonary hypertension and a fixed low CO. The cause is inadequate cerebral blood flow due to a combination of failure to increase the CO, along with diversion of systemic blood flow toward the exercising muscles. Syncope may also occur at rest in association with the onset of bradycardia, presumably vagal in origin. Hoarseness, due to paralysis of the left recurrent laryngeal nerve, may result from trapping of the nerve between the aorta and the dilated left pulmonary artery (a form of Ortner syndrome). If the right ventricle should fail, lower extremity swelling is common, as are abdominal signs and symptoms including a sensation of “bloating,” early satiety, tender hepatomegaly, ascites, and abdominal pain. Symptoms of right ventricular failure and the presence of syncopal events are associated with a worse prognosis. Hemoptysis in the setting of pulmonary hypertension is most often due to pulmonary venous congestion but when mitral stenosis is present, it is generally attributed to bleeding from bronchial veins. Occasionally, hemoptysis occurs in other forms of pulmonary hypertension and may originate in alveolar capillaries, precapillaries, or elsewhere in the pulmonary arterial tree.
Not infrequently, the suspicion of pulmonary hypertension is raised by the presence of a known risk for pulmonary hypertension (e.g., systemic sclerosis) or by serendipitous discovery of right ventricular enlargement by an electrocardiogram or chest radiograph taken for other reasons (Figs. 72-5 and 72-6). Initial recognition of the presence of pulmonary hypertension also frequently occurs in the absence of reported symptoms when an echocardiogram is performed for the evaluation of a murmur. Alternatively, echocardiographic evidence of pulmonary hypertension may be found unsuspectedly when the study is obtained as “routine” evaluation of a patient complaining of any of a number of chest symptoms, including dyspnea. Patients with severe pulmonary hypertension are prone to sudden death and its occurrence may be the first (and last) indication of disease. Death has occurred unexpectedly during normal activities, cardiac catheterization, and surgical procedures, and after the administration of sedatives or anesthetic agents. In a few instances, bradycardia leading to cardiac arrest has preceded sudden death.
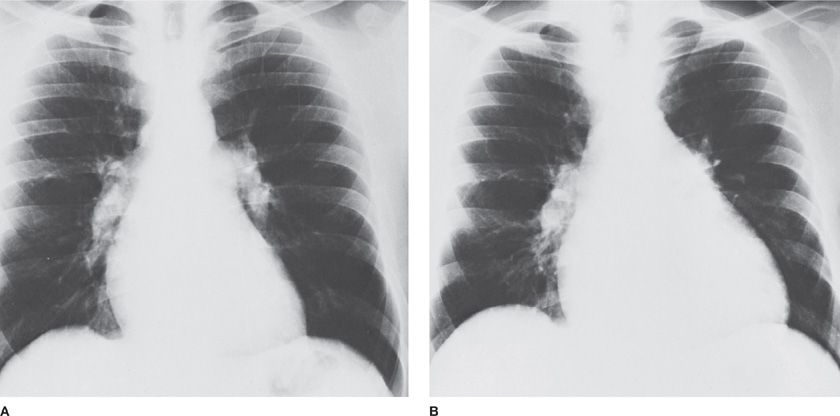
Figure 72-5 Radiographic changes in idiopathic pulmonary arterial hypertension. As compared to a chest radiograph 14 months earlier (A) enlargement of the cardiac silhouette has occurred in a 30-year-old man in association with increasing dyspnea (B). Decrease in the cardiac silhouette occurred in response to chronic pulmonary vasodilator therapy.
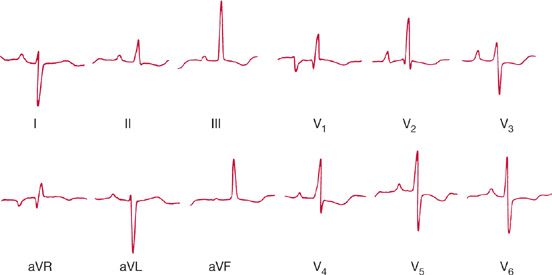
Figure 72-6 Twenty-six-year-old woman in whom the first evidence of primary pulmonary hypertension was by electrocardiography. The record shows marked right axis deviation and dominant R waves over the right precordium consistent with right ventricular hypertrophy.
An important hint to the presence of pulmonary hypertension is a history of long-standing dyspnea that has not responded to treatment for other more common disorders. Patients have often seen several clinicians before an appropriate diagnosis is made. Younger patients have frequently been told their symptoms are due to asthma, yet failed to improve significantly with aggressive anti-inflammatory and bronchodilator therapies. In older patients, dyspnea or exercise limitation may have been blamed on COPD, often despite a trivial history of tobacco use. Unfortunately, a further clue in such patients is learning that these “diagnostic” labels were never investigated with pulmonary function testing. Patients should be asked about important symptoms or risk factors that might suggest the presence and cause of pulmonary hypertension. These include symptoms of collagen vascular disease (e.g., dysphagia, skin or joint changes, Raynaud’s phenomenon), sleep apnea (e.g., witnessed apneic events, daytime hypersomnolence), history of risks for thromboembolism, HIV infection, liver disease, or anorectic agent use. A history of tobacco use and chronic sputum production, or a known history of asthma with poor control might be important clues to the presence of obstructive airway disease and hypoxia as the cause of pulmonary hypertension. A prior history of interstitial lung disease or any cause of chronic hypoxia must be noted. A careful family history should be taken including inquiring about relatives who suffer(ed) poorly understood cardiopulmonary conditions.
 PHYSICAL EXAMINATION
PHYSICAL EXAMINATION
A careful physical examination is necessary to recognize the presence of pulmonary hypertension and begin elucidating its cause (Table 72-5). In mild-to-moderate pulmonary hypertension, the physical examination is apt to be unrevealing unless suspicion has been aroused that pulmonary hypertension may be present. Right ventricular enlargement is an important clue but notoriously difficult to detect in its early stages on physical examination. Evidence of pulmonary hypertension, such as prominent closure of the pulmonary valve, is apt to be overlooked or discounted, especially in younger people; recognition of tricuspid insufficiency or a right ventricular gallop is often delayed until pulmonary hypertension becomes severe and has led to heart failure.



