Chapter 56 Pulmonary Arterial Hypertension
Definition and Classification of Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is formally defined as a mean pulmonary artery pressure (PAP) of 25 mmHg or greater at rest or 30 mmHg or greater during exercise. This is accompanied by a pulmonary vascular resistance (PVR) of greater than 3 Wood units (WU), with a normal pulmonary artery wedge pressure (< 15 mmHg) in the absence of any known cause of pulmonary hypertension (PH). Pulmonary arterial hypertension encompasses one of the five categories of PH, based on the most recent classification system.1 The pathophysiology and clinical characteristics of categories 2 through 5 of PH (i.e., PH associated with left-sided heart disease; lung disease/chronic hypoxia; thromboembolic PH; and other groups) are reviewed in Chapter 57. In this chapter, we will focus on category 1, PAH, which includes idiopathic and familial PAH as well as PAH associated with various other diseases (Box 56-1). Until improved therapeutic options became available during the past 2 decades, PAH was considered a rare but rapidly progressive and devastating condition, leading to death from right heart failure by a median of 2.8 years from the time of diagnosis.2 In recent years, an improved conceptual framework has been developed to better delineate the pathogenesis and clinical presentation of PAH, accompanied by improved directed therapeutic approaches. Nonetheless, PAH still carries a 15% annual mortality rate, and current therapeutics have been mostly effective at slowing illness progression rather than reversing or curing the disease. This chapter will highlight current molecular understanding of the complex disease, via the still incompletely characterized interplay of genetic and exogenous upstream stimuli with downstream vascular effectors. This framework will then be coupled to current understanding of the clinical presentation and progression of PAH and existing and evolving treatment modalities.
 Box 56-1 Clinical Classification of Pulmonary Arterial Hypertension
Box 56-1 Clinical Classification of Pulmonary Arterial Hypertension
Associated with significant venous or capillary involvement:
Adapted with permission from Simmoneau G, Galiè N, Rubin L, et al: Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43:5 S–12 S, 2004.
Epidemiology
Early epidemiological data from the 1980s estimated the incidence of PAH at one to two cases per million people in the general U.S. population.2 More recent estimates in Europe3 place the incidence and prevalence of PAH, respectively, at 2.4 cases per million annually and 15 cases per million in France and 7.6 cases per million annually and 26 cases per million in Scotland. Globally, the prevalence may be much higher given that many risk factors for PAH, such as human immunodeficiency virus (HIV) and schistosomiasis, are also more prevalent in the developing world. An exact number is difficult to estimate on a global scale, given the difficulty of diagnosing PAH and overall limited access to health care worldwide.4 The disease affects women more frequently than men (1.7:1). This female predominance is exaggerated in the population of African descent (4.3:1), although the overall racial distribution of patients reflects that in the general population.
Primary PAH presents most commonly in the fourth decade of life; ages range from 1 to 81, with 9% of patients older than 60 years of age.5 Some sources cite a similar gender ratio among children diagnosed with the disease, whereas others note an equal distribution between male and female children.6 Primary PAH is typically difficult to diagnose, and the average time from onset of symptoms to diagnosis was 2 years in the National Institutes of Health (NIH) Registry.2 More recently, however, thanks to improved clinical awareness and more sophisticated invasive and noninvasive techniques for PAP measurements, time to diagnosis has decreased considerably, at least in the developed world.
Molecular Pathogenesis of Pulmonary Arterial Hypertension
In humans, the natural history of PAH lesions is unknown because patients usually present when the disease is advanced. The pathological appearance of severe PAH is similar regardless of cause and reflects the end stage of a common response to pulmonary vascular injury. Common histological features in nearly all types of PAH occur at the level of the small peripheral pulmonary arteries (100-1000 μm) (Fig. 56-1); these include intimal fibrosis, distal localization and proliferation of vascular smooth muscle, and pulmonary arterial occlusion.7 A hallmark of severe end-stage disease is the formation of a vessel “neointima,” characterized by increased deposition of extracellular matrix (ECM) and myofibroblasts. Plexiform lesions can predominate, characterized by over-proliferation of endothelial-like cells encroaching upon the vessel lumen.

Figure 56-1 Histopathology of pulmonary arterial hypertension (PAH).
(Courtesy JL Faul, MD, Stanford University.)
Multiple cell types in the pulmonary arterial wall and pulmonary arterial circulation contribute to the specific response to injury and development of vessel remodeling8 (Fig. 56-2). The endothelium serves as a central sensor of injurious stimuli such as hypoxia, shear stress, inflammation, and toxins. Dysregulation of numerous downstream vascular effectors may be the result of initial endothelial cell (EC) injury or dysfunction. It has been hypothesized that endothelial apoptosis early in PAH initiates selection of apoptosis-resistant ECs that can further proliferate via monoclonal amplification in plexiform populations. This has led to speculation of a model of end-stage PAH similar to that of progression to cancer, with dysregulation of the cell cycle and apoptosis as predominant features. Similarly, dysregulated growth of pulmonary artery smooth muscle cells (SMCs) also plays a key role in PAH progression because apoptosis is suppressed while proliferation increases.
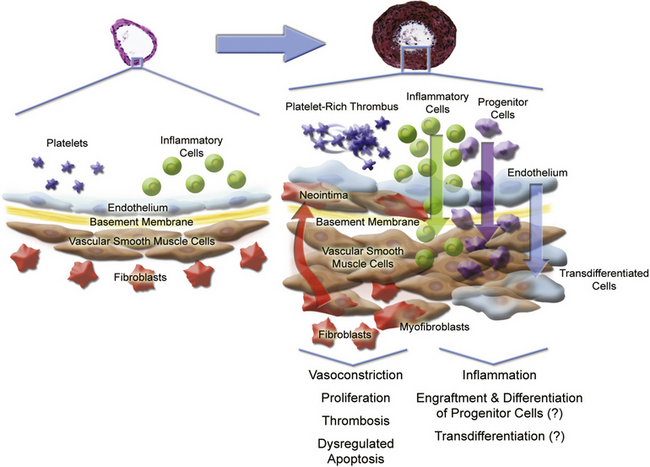
Figure 56-2 Pulmonary arterial pathobiology involves coordinate action of multiple cell types.
(Adapted with permission from Chan S, Loscalzo J: Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol 44:14–30, 2008. Micrographs of pulmonary arteries courtesy www.scleroderma.org and Humbert M, et al: Treatment of pulmonary arterial hypertension. N Engl J Med, 2004, 351:1425–1436, 2004. Copyright 2004, Massachusetts Medical Society. All rights reserved.)
Genetic Association
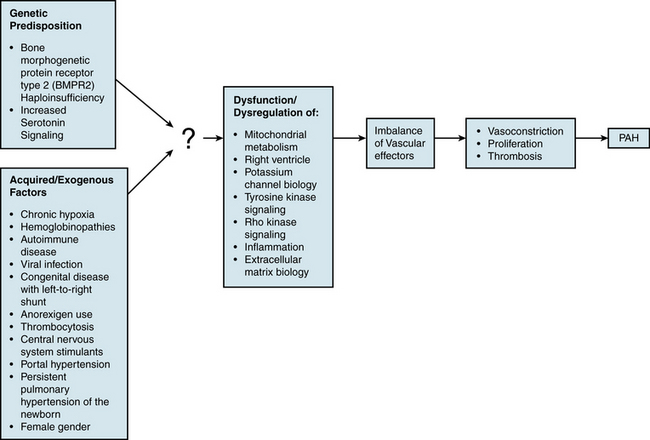
An understanding of the mechanism of genetic predisposition to PAH is of paramount importance for identifying the root pathogenesis (Fig. 56-3). The familial variety of idiopathic PAH accounts for at least 6% of all cases of PAH.7 Pedigree analysis has demonstrated an autosomal dominant inheritance but with variable penetrance; only 10% to 20% of putative genetic carriers develop clinical PAH. Genetic anticipation is present, since each successive generation of affected families is afflicted at a younger age and greater severity compared with the preceding generation.
Bone morphogenetic receptor-II
Mutations in the transforming growth factor (TGF)-β receptor superfamily have been genetically linked to PAH and likely play a causative role in disease development. A rare group of patients with hereditary hemorrhagic telangiectasia (HHT) and idiopathic PAH harbor specific mutations in ALK1 or endoglin, genes encoding for two such members of the TGF-β receptor superfamily.9,10 However, a more prevalent cohort of PAH patients carries mutations in the bone morphogenetic protein receptor type II (BMPR2 gene which encodes for BMPR2).11,12 Over 140 mutations in BMPR2 have been reported in patients with familial PAH,13 mainly located in the extracellular ligand-binding domain, cytoplasmic serine/threonine kinase domain, or long carboxyterminal domain. These account for 70% of all familial pedigrees of PAH and 10% to 30% of idiopathic PAH cases.8 BMPR2 loss-of-function mutations have only been found in the heterozygous state. The absence of BMPR2 mutations in some familial cohorts and in most sporadic cases indicates that additional unidentified genetic mutations can also predispose to development of PAH. Furthermore, the presence of incomplete penetrance (approximately 25% of carriers in affected families develop clinical PAH) and genetic anticipation suggests that BMPR2 mutations are necessary but insufficient alone to result in clinically significant disease.
The mechanism of action of BMPR2 is complex, and its role in PAH progression is still unclear (Fig. 56-4A). It functions as a receptor with serine/threonine kinase activity, and it activates a broad and complex range of intracellular signaling pathways (as reviewed in14). Upon binding one of many possible bone morphogenetic protein (BMP) ligands, BMPR2 forms a heterodimer with one of three type-I receptors. BMPR2 phosphorylates the bound type-I receptor, which in turn phosphorylates one of the Smad family of proteins to allow for nuclear translocation, binding to deoxyribonucleic acid (DNA), and regulation of gene transcription. Alternatively, BMPR2 activation can also lead to signaling via the LIM kinase pathway, p38/ mitogen-activated protein kinase/extracellular signal regulated kinase/c-jun NH 2-terminal kinase (MAPK/ERK/JNK) pathways, or c-Src pathway, independent of Smad activation.
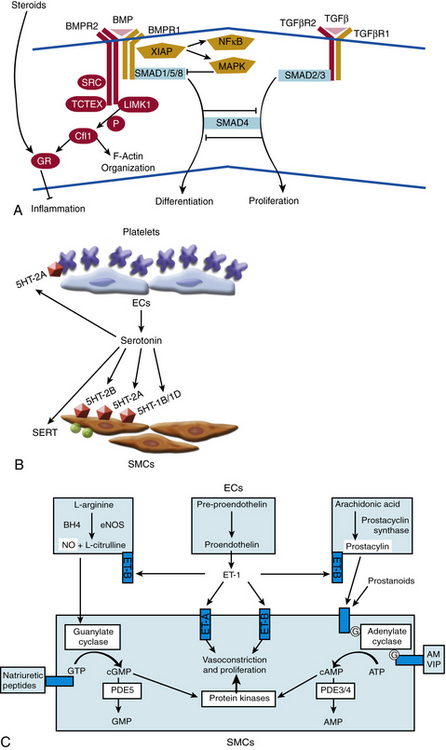
Figure 56-4 Molecular signaling mechanisms of pulmonary arterial hypertension (PAH).
(Adapted from Archer SL, Weir EK, Wilkins MR: Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121:2045–2066, 2010.)
Cellular effects of BMPR2 activation are multiform. In the adult, BMPR2 is expressed predominantly in pulmonary endothelium, medial SMCs, and macrophages.15 Under normal conditions, BMP ligands bind BMPR2 to suppress growth of VSMCs. In contrast, binding of BMP2 and BMP7 to BMPR2 in pulmonary endothelium protects against apoptosis. A widely held hypothesis contends that failure of the suppressive effects of BMP ligands on vascular smooth muscle and failure of the protective effects of BMP ligands on endothelium may trigger vascular proliferation and remodeling. Accordingly, in VSMCs derived from patients with familial PAH harboring BMPR2 mutations, exposure of BMP ligands does not suppress proliferation. Unlike the response in wild-type endothelium, exposure of ECs cultured from patients with idiopathic PAH to BMP2 does not protect against apoptosis.16 These dysfunctional signaling pathways have been corroborated in some rodent models of PAH. In correlation, pulmonary levels of BMPR2 are reduced both in familial cases of PAH without any BMPR2 mutation and in cases of secondary PAH.15 Thus, dysregulation of the BMP signaling pathway may be a common pathogenic finding in multiple types of PAH due to genetic or exogenous stimuli, but the definitive in vivo effects of these mutations have been difficult to decipher. Specifically, mouse models harboring specific BMPR2 heterozygous mutations have failed to exhibit robust PAH under static conditions,17,18 again suggesting that dysfunctional BMPR2 is likely insufficient alone to cause disease. As a result, a clear mechanistic explanation of the impact of BMPR2 mutations on pathogenesis remains elusive.
Additional genetic pathways
In addition to BMPR2 haploinsufficiency, alternative mechanisms involving complementary “modifier” genes may also contribute to a genetic predisposition to PAH. The most promising data identifying such modifier genes have analyzed the association of particular single nucleotide polymorphisms (SNPs) with the development of PAH. Thus far, SNP variants have suggested certain genes such as the serotonin transporter (SERT), Kv1.5, and the trp cation channel, subfamily C, member 6 (TRPC6).3 Such associations do not always suggest a causal relationship, so additional mechanistic data are necessary for proper interpretation.
In the case of SERT and serotonin signaling, supportive data are more prevalent. Serotonin is both a vasoconstrictor and mitogen that promotes smooth muscle hyperplasia and hypertrophy (see Fig. 56-4B). Primarily stored in platelet granules, secreted serotonin binds G protein–coupled serotonin receptors (GPCRs) present on pulmonary artery SMCs. Activation of these receptors leads to a decrease in adenylyl cyclase and cyclic adenosine monophosphate (cAMP), resulting in increased contraction. Furthermore, the cell-surface SERT allows for transport of extracellular serotonin into the cytoplasm of SMCs, thereby activating cellular proliferation directly through the action of serotonin or indirectly via potential pleiotropic mechanisms.
A number of observations in idiopathic and familial disease, congenital disease, and environmental exposure have implicated the proliferative effects of serotonin in PAH.19 In idiopathic PAH, pulmonary expression of serotonin receptors is increased, and plasma levels of serotonin are chronically elevated. A mouse model of hypoxic PH parallels these changes.20 A positive association has been noted among patients with congenital platelet defects in serotonin uptake (i.e., delta storage pool disease) and development of PAH. Chronic exposure to anorexigens, such dexfenfluramine (an inhibitor of serotonin reuptake and stimulator of serotonin secretion), leads to increased levels of circulating free serotonin. In mice, these changes are accompanied by increased 5HT receptor type 2B response and inhibition of SERT responses. In humans, these changes correlate with an increased risk for development of PAH. In addition, the L-allelic variant of SERT is associated with increased expression of the transporter and enhanced smooth muscle proliferation. In some human studies,21 this variant has been associated with an increased risk of PAH in the homozygous population.
Animal models of PH have also implicated the activated serotonin pathway in disease progression. Treatment with serotonin and chronic hypoxia in a rat model led to worsened hemodynamics and increased vessel remodeling.22 Exposure to increased serotonin led to worsened PH in a BMPR2+/− heterozygote murine model.23 Similarly, overexpression of SERT in mice resulted in spontaneous development of PAH in the absence of hypoxia and exaggeration of PH after a hypoxic stimulus.24 Conversely, vessel remodeling and hypoxic PH are reduced in a 5HT1B receptor–null mouse25 and are abrogated in a 5HT2B receptor–null mouse.20 As a result, serotonin signaling modulates pulmonary smooth muscle function in both normal and disease states and likely contributes to disease progression of PAH. However, attempts at using selective serotonin reuptake inhibitors (SSRIs) as a therapeutic approach have yielded mixed results3 to date. The exact contribution of this mechanism in PAH requires further clarification.
Acquired/Exogenous Factors
In addition to genetic predisposition, development of PAH likely depends on a variety of physiological, acquired, and/or exogenous stimuli. Some of these factors have been studied to a sufficient degree to hypothesize potential pathogenic mechanisms (see Fig. 56-3).
Chronic hypoxia
Pulmonary vascular response to hypoxia has been well studied in cell culture and animal models,26 but its impact on PAH is unclear. In general, pulmonary vascular responses in acute and chronic hypoxia likely allow for the propagation of PAH, and therefore may contribute to later stages of the disease. Acute hypoxia induces vasodilation in systemic vessels but induces vasoconstriction in pulmonary arteries. This acute and reversible effect is mediated in part by up-regulation of endothelin-1 (ET-1) and serotonin, and in part by hypoxia- and redox-sensitive potassium channel activity in pulmonary VSMCs. Coordinately, these events lead to membrane depolarization in SMCs, increase in cytosolic calcium, and vasoconstriction.27 In contrast, chronic hypoxia induces vascular remodeling and less reversible changes, including migration and proliferation of VSMCs and deposition of ECM. These cellular events in chronic hypoxia correlate with the remodeling events inend-stage PAH; however, because the histopathology of hypoxic PH does not recapitulate all aspects of PAH, some mechanistic differences in pathogenesis certainly exist but have not yet been fully identified. These are important considerations, especially in the context of interpreting studies of hypoxic PH and extrapolating those findings to the pathogenesis of PAH.
Hemoglobin disorders
Pulmonary arterial hypertension is associated with hemoglobinopathies, especially thalassemias and sickle cell anemia.28 Hemolysis accompanying these disorders may lead to destruction of bioactive nitric oxide (NO) by free hemoglobin or reactive oxygen species (ROS). Furthermore, ROS may lead to increased levels of oxyhemoglobin, which further impairs delivery of NO to the vessel wall. As a result of the lack of available NO, an inflammatory and proliferative cascade may ensue, with culmination in PAH. Accordingly, decreased NO bioavailability with development of PH has been reported after hemolysis in a murine model of sickle cell disease (SCD),29 and acutely in a murine model of intravascular hemolysis.30 In vivo correlation to human disease is pending.
Portopulmonary hypertension
Portopulmonary hypertension is defined as PAH associated with portal hypertension (portal pressure >10 mmHg), with or without hepatic disease.31 Approximately 9% of patients with severe PAH are reported to have portal hypertension. Portopulmonary hypertension affects 4% to 6% of patients referred for liver transplantation. Liver transplantation perioperative mortality is significantly increased in patients with a mean PAP above 35 mmHg. Diagnosis of PH is usually made 4 to 7 years after the diagnosis of portal hypertension, but it has occasionally been reported to precede onset of portal hypertension. Risk of PH increases with duration of disease. The correlation between severity of portal hypertension and development of PH is debated. The female predominance of primary PAH is not seen in portopulmonary hypertension.
Collagen vascular disease
Pulmonary arteriopathy complicates autoimmune diseases, especially in the setting of the CREST (calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasia) variant of limited systemic sclerosis and, to a lesser degree, in mixed connective tissue disease, systemic lupus erythematosus (SLE), and rheumatoid arthritis.32 Pulmonary arterial hypertension has been reported in approximately 10% to 30% of patients with mixed connective tissue disease, 5% to 10% of patients with SLE, and more rarely in the settings of rheumatoid arthritis, dermatomyositis, and polymyositis.33 Sjögren syndrome may rarely be complicated by rapidly progressive PAH. It is particularly important to distinguish between PAH and thromboembolic PH in patients with SLE and antiphospholipid syndrome. Occurrence of PAH in each disease has been associated with Raynaud phenomenon, suggesting at least some similarities in pathogenesis.34 Presence of interstitial lung disease and pulmonary fibrosis, seen at varying frequency in these autoimmune syndromes, may represent a common pathogenic factor in development of PAH. Accordingly, in the setting of pulmonary fibrosis and hypoxia, significant perivascular inflammation and deposition of ECM have been observed, which may increase vasoconstriction, proliferation, and vessel remodeling. Murine models of interstitial lung disease may prove important in further elucidating pathogenic mechanisms.
Human immunodeficiency virus
An association between HIV infection and PH has been noted in approximately 0.5% of all patients with HIV infection, a rate 6 to 12 times higher than the general population.35 Notably, HIV does not infect pulmonary arterial endothelium, but mechanisms of disease have been proposed that directly stem from effects of HIV infection.36 These include infection of SMCs with subsequent dysregulation of proliferation, imbalance of vascular mitogens in response to systemic HIV infection, and endothelial injury precipitated by HIV-infected T cells. The direct actions of HIV-encoded proteins may also factor into PAH development.8 The HIV gp120 protein may induce pulmonary endothelial dysfunction and apoptosis. In a macaque model of simian immunodeficiency virus infection, a pathogenic interaction of the viral Nef protein with the pulmonary vessel wall has been reported, leading to pulmonary arteriopathy. Cell culture studies have also demonstrated a role for the HIV Tat protein in repression of BMPR2 transcription, potentially provoking a proliferative response in the vessel wall.
It had been proposed that human herpesvirus 8 (HHV-8), the causative agent of Kaposi sarcoma and an opportunistic pathogen highly associated with HIV infection, may play a role in PAH development with progression to plexiform lesions. Although it was initially reported that HHV-8 infection is associated with idiopathic PAH,37 that link has not been consistently validated after study of additional populations. Nonetheless, PAH in the setting of HIV infection likely results from multifactorial effects, and the underlying pathogenesis may involve both direct results of viral infection and indirect consequences of associated pathogens.
Shunt physiology
Increased flow through the pulmonary circulation has long been associated with development of PAH. Certain types of congenital heart disease with functional systemic-to-pulmonary shunts, such as unrestricted ventricular septal defects (VSDs) and large patent ductus arteriosus (PDA), invariably lead to pulmonary vascular remodeling and the clinical syndrome of PAH during childhood (Eisenmenger’s syndrome).38 Approximately a third of patients with uncorrected VSDs and PDAs die from PAH. The timing of surgical repair greatly influences the outcome. If the shunt is repaired within the first 8 months of life, patients tend to have normal pulmonary pressures regardless of pathological findings; by contrast, patients operated on after age 2 tend to have persistent PAH. Importantly, when PVR equals or exceeds systemic vascular resistance, surgical correction of a shunt will increase the load on an already overburdened right ventricle (RV), worsen the patient’s clinical condition, and not reverse PAH.
The presence of atrial septal defects (ASD) with systemic-to-pulmonary shunts may also lead to PAH over time.39 Yet, in contrast to cases of unrestricted VSD and PDA, only 10% to 20% of all persons with ASDs progress to PAH. This observation may reflect differences in the response of the pulmonary vasculature to pressure overload (as seen in shunts with VSD and PDA) as compared to volume overload (as seen in shunts with ASD). Furthermore, patients with ASD may harbor a specific unidentified genetic predisposition to the development of PAH that may exacerbate the increased volume load to the pulmonary circulation.
At the molecular level, the physiological flow patterns of laminar shear stress, turbulent flow, and cyclic strain are all recognized by ECs, leading to transduction of intracellular signals and modulation of a wide variety of phenotypic changes.40 Significant prior work has focused mainly on the endothelium of the peripheral vasculature, suggesting that laminar flow induces a vasoprotective quiescent vascular state, whereas turbulent flow leads to a proinflammatory and thrombogenic state. It is unclear whether these flow-dependent phenotypes are recapitulated in the pulmonary vasculature. In part, this stems from the difficulty of directly studying directly the in vivo flow patterns at the anatomical level of the pulmonary arteriole. Ex vivo modeling of pulsatile flow with high levels of shear stress and chronic vascular endothelial growth factor (VEGF) inhibition has demonstrated apoptosis of pulmonary artery ECs, followed by outgrowth and selection for proliferating apoptosis-resistant cells.41 Therefore, chronically elevated flow may allow for selection of cells with dysregulated EC growth and resulting clonal or polyclonal expansion to plexiform lesions.
Persistent pulmonary hypertension of the newborn
Persistent pulmonary hypertension of the newborn (PPHN) is characterized by persistent elevation of PVR, right-to-left shunting, and severe hypoxemia. It can occur with pulmonary parenchymal diseases including sepsis, meconium aspiration, pneumonia, maladaptation of the pulmonary vascular bed, or without an apparent cause.42 Persistent PH in newborns may lead to death during the neonatal period, or it may be transient, leading to spontaneous and complete recovery. Inadequate production of NO may be an important contributor to persistent PH in infants, and inhaled NO is useful in treating this disorder.
Miscellaneous disorders
Pulmonary arterial hypertension is more rarely reported in patients suffering from other clinical syndromes such as pulmonary veno-occlusive disease, chronic myelodysplastic syndromes with thrombocytosis, and idiopathic thrombocythemia, as well as in persons exposed to stimulants of the central nervous system, such as methamphetamines and cocaine. Dysregulation of serotoninergic signaling may contribute8 but does not explain these associations entirely. Finally, idiopathic PAH demonstrates a gender predilection of unclear etiology, with a high predominance of affected females.43
Vascular Effectors
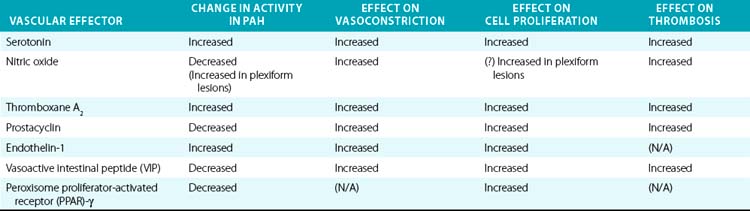
Downstream of the genetic and acquired triggers of PAH, the histopathological processes that predominate later stages of disease include vasoconstriction, cellular proliferation, and thrombosis. These processes are influenced by a complex and dysregulated balance of vascular effectors controlling vasodilation and vasoconstriction, growth suppressors and growth factors, and pro- vs. antithrombotic mediators. Most of these effectors have been described in previous comprehensive reviews and will be described here specifically in regard to their known roles in PAH pathogenesis (Table 56-1).
Nitric oxide
Gaseous vasoactive molecules regulate pulmonary vascular homeostasis, and alterations in their endogenous production have been linked to progression of PAH. Nitric oxide is the best described of these factors (see Fig. 56-4C).44 It is a potent pulmonary arterial vasodilator as well as a direct inhibitor of platelet activation and VSMC proliferation. Nitric oxide diffuses into recipient cells (e.g., vascular muscle), where it activates soluble guanylyl cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP). In turn, cGMP interacts with at least three different groups of effectors: cGMP-dependent protein kinases, cGMP-regulated phosphodiesterase (PDE), and cyclic nucleotide-gated ion channels. Synthesis of NO is mediated by a family of NO synthase (NOS) enzymes. In the pulmonary vasculature, the endothelial (eNOS) isoform figures most prominently and is regulated by a multitude of vasoactive factors and physiological stimuli including hypoxia, inflammation, and oxidative stress. Reduced levels and reduced activity of eNOS, reduced NO bioavailability, dysregulated intracellular NO transport via caveolae, and increased degradation of cGMP all aggravate PAH progression in human studies and/or animal models.
Dysregulation of NO may also depend upon still incompletely characterized processes of NO transport in blood.45 Specific polymorphisms of NOS have also been associated with pulmonary hypertension. Together, these effects indicate a coordinated mechanism of dysregulated vasoconstriction. Correspondingly, murine models that genetically lack eNOS, GTP cyclohydrase-1 (the rate limiting enzyme for synthesis of an essential cofactor of NOS, tetrahydrobiopterin), or dimethylarginine dimethylaminohydrolase (DDAH, an enzyme important in degradation of NOS inhibitors) all display exaggerated susceptibility to developing PH in response to other endogenous stimuli.3 Finally, the impact of NO has been reflected in its therapeutic role in PAH, such as inhaled NO46 and the NO-dependent phosphodiesterase type-5 (PDE5) inhibitors (discussed later in detail).
Prostacyclin/thromboxanes
The arachidonic acid metabolites prostacyclin and thromboxane A2 (TxA2) also play crucial roles in vasoconstriction, thrombosis, and to a certain degree, vessel wall proliferation (see Fig. 56-4C). Prostacyclin (prostaglandin I2) activates cAMP-dependent pathways and serves as a vasodilator, antiproliferative agent for vascular smooth muscle, and inhibitor of platelet activation and aggregation. In contrast, TxA2 increases vasoconstriction and activates platelets.47 Protein levels of prostacyclin synthase are decreased in small and medium-sized pulmonary arteries in patients with PAH, particularly with the idiopathic form.48 Biochemical analysis of urine in patients with PAH has shown decreased levels of a breakdown product of prostacyclin (6-ketoprostacyclin F2α), accompanied by increased levels of a metabolite of TxA2 (thromboxane B2).49 Therefore, it appears that production of these effectors is coordinately regulated, with the imbalance toward TxA2 favored in the development of PAH. The mode of this regulation remains to be characterized. Nonetheless, recognition of this imbalance has led to the success of prostacyclin therapy and improvement of hemodynamics, clinical status, and survival in patients with severe PAH.50
Endothelin-1
Endothelin-1 is expressed by pulmonary ECs and has been identified as a significant vascular mediator in PAH (see Fig. 56-4C).51 It acts as both a potent pulmonary arterial vasoconstrictor and mitogen of pulmonary smooth muscle cells. The vasoconstrictor response relies upon binding to the endothelin receptor A (ET-A) on vascular smooth muscle cells. This leads to an increase in intracellular calcium, along with activation of protein kinase C (PKC), mitogen-activated protein kinase (MAPK), and the early growth response genes c-fos and c-jun.52 The mitogenic action of ET-1 on pulmonary VSMCs can occur through either the ET-A and/or the ET-B receptor subtype, depending on the anatomical location of cells. Endothelin receptor A predominantly mediates mitogenesis in the main pulmonary artery, whereas mitogenesis in resistance arteries relies upon contributions from both subtypes. The resulting vasoconstriction, mitogenesis, and vascular remodeling are thought to lead to significant hemodynamic changes in the pulmonary vasculature and to PAH. Plasma levels of ET-1 are increased in animal and human subjects suffering from PAH due to a variety of etiologies53 and correlate with disease severity.54 Again, improvement in hemodynamics, clinical status, and survival of PAH patients treated with chronic ET receptor antagonists highlights the significance of these effects.55
Vasoactive intestinal peptide
Down-regulation of vasoactive intestinal peptide (VIP) may also play a pathogenic role. Vasoactive intestinal peptide is a pulmonary vasodilator, an inhibitor of proliferation of VSMCs, an inhibitor of platelet aggregation, and free radical scavenger. Decreased concentrations of VIP and VIP receptors have been reported in serum and lung tissue of patients with PAH.56 Vasoactive intestinal peptide-null mice suffer from moderate pulmonary hypertension.57 Both pulmonary arterial pressure (PAP) and PVR in humans decrease after treatment with VIP.58,59 Key questions regarding the mode(s) of regulation of VIP expression and its putative causative role in PAH remain unanswered.
Peroxisome proliferator-activated receptor γ
Peroxisome proliferator-activated receptor gamma (PPAR-γ)and apolipoprotein E (apoE) may function as integral factors in PAH.60,61 PPARs are ligand-activated nuclear transcription factors that heterodimerize with the retinoid X receptor for subsequent binding to PPAR response elements in the promoters of target genes. Idiopathic PAH patients carry reduced pulmonary transcript levels of PPAR-γ and apoE. PPAR-γ in particular is a direct target of BMP2 in human PASMCs, leading to stimulation of apoE synthesis and downstream inhibition of vascular smooth muscle proliferation. PPAR-γ also regulates a host of protein kinases implicated in PASMC proliferation and migration. PPAR-γ agonists are antiinflammatory and induce pro-apoptotic phenotypes, both theoretically inhibitory to the pathogenesis of PAH. Mice deficient in smooth muscle–specific PPAR-γ are prone to PAH. Similarly, when fed a high-fat diet, male mice deficient in apoE develop PAH. This condition is reversed by rosiglitazone, a PPAR-γ agonist. Nonetheless, less robust results have been seen when using rosiglitazone hypoxic-PH rats, suggesting that these results offer a partial explanation to the complex molecular pathogenesis of BMPR2 signaling and PAH pathogenesis.
Natriuretic peptides
The atrial and brain natriuretic peptides (ANP and BNP) are produced by myocardium in response to stretch. They bind guanylyl cyclase receptors (NPR-A and NPR-B) that induce production of cGMP. Increased plasma concentrations of these peptides in PAH have been used as markers of the extent of RV dysfunction.62 Genetic inactivation of NPR-A in mice is associated with PH63; in contrast, administration of atrial natriuretic peptide (ANP) ameliorates PAH in rodent models.64 Additionally, inhibition of the metabolic breakdown of natriuretic peptides via neutral endopeptidase inhibitors has shown promise in animal studies of PAH. Human studies have yet to confirm efficacy.
Adrenomedullin
Adrenomedullin is an endogenous peptide that activates signaling pathways (i.e., cAMP, NO-cGMP, phosphatidylinositol-3-kinase/Akt) to induce vasodilation and inhibit proliferation. It decreases mean PAP and RV hypertrophy in hypoxic rats.65 A related peptide, adrenomedullin-2, binds the same cellular receptors as adrenomedullin, and it is elevated in some rodent forms of PAH.
Miscellaneous effectors
Other potential contributing factors include angiopoietin-1, the vasodilatory gases carbon monoxide and hydrogen sulfide, phosphodiesterase I, survivin, the calcium binding protein S100A4/Mts, the transient receptor potential channels, and Notch 3.66 These may represent important but as yet incompletely described pathogenic contributors.8 Other mitogenic and angiogenic growth factors, such as VEGF, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF)-2, insulin-like growth factor (IGF)-1, and epidermal growth factor (EGF) all may play downstream roles in later stages of PAH.
Pathogenic Pathways
Our understanding of specific actions of individual effectors has improved, but how they relate to upstream genetic or exogenous triggers of PAH remains unclear. In fact, none of these factors has yet been definitively linked to the root pathogenesis of disease. Insight into this topic is offered by the fact that known effectors are likely subject to upstream, overarching regulatory pathways that affect the action of multiple vasoactive molecules.67 Characterization of these regulatory mechanisms may eventually allow for identification of primary molecular triggers of disease and offer novel therapeutic targets for drug development (see Fig. 56-3).
Mitochondrial metabolism
Mitochondrial and metabolic dysfunction may be common in PAH. Endothelial and VSMCs from human PAH tissue display dysmorphic and hyperpolarized mitochondria.68 Metabolically, these cells preferentially exhibit a down-regulation of mitochondrial metabolism with an induction of glycolysis for energy production.69 Under hypoxic conditions, this so-called Pasteur effect is a normal adaptive event that improves cellular survival by optimizing adenosine triphosphate (ATP) production while reducing oxygen radicals generated from the mitochondrial electron transport chain. Under normoxic conditions, however, such a shift to glycolysis (the Warburg effect) is thought to confer inappropriate resistance to apoptosis and is prominently seen in various cancer lineages. Such metabolic changes in the mitochondria are dependent upon a master transcription factor of hypoxia, hypoxia inducible factor 1 alpha (HIF-1α), and its critical downstream targets such as pyruvate dehydrogenase kinase (PDK1), among others.70 In PAH, HIF-1α and PDK1 are dysregulated; conversely, treatment with dichloroacetate, a PDK inhibitor, ameliorates multiple forms of PAH in rodent models (chronic hypoxic PH, monocrotaline PAH, and fawn-hooded rat PAH).71 It is conceivable that therapies aimed at reversing this metabolic dysregulation may result in improvement and/or regression of the PAH phenotype.
Right ventricular dysfunction
After birth, PAPs typically decline, leading to involution of the RV to a thin-walled structure in the adult. During pathological conditions of increased PAPs in PAH, however, RV hypertrophy (RVH) and strain ensue, followed by RV failure if left untreated. Historically, the molecular pathways governing left ventricular (LV) failure, which have been better characterized, had been assumed to play primary roles in RV failure as well. Contemporary evidence suggests there may be distinct molecular and physiological differences between LV and RV failure that are just beginning to be explored.72 For instance, PDE5, which is expressed in the fetal RV, is selectively up-regulated in the hypertrophied RV. Interestingly, inhibition of PDE5 enhances RV contractility without affecting the LV, where PDE5 expression is absent.
Unique metabolic changes in the RV have also been reported. The normal RV can use either fatty acids or glucose as needed for energy production. In RVH, the RV nearly exclusively relies upon glycolysis. Adenosine monophosphate (AMP)-activated protein kinase is activated in RVH, which preserves ATP levels by increasing glucose transport and glycolysis while inhibiting the tricarboxylic acid (TCA) cycle via repression of acetyl-coenzyme A carboxylase.73 Furthermore, during RVH, PDK is activated, inducing a metabolic switch away from oxidative phosphorylation to glycolysis. As a result, the hypertrophied RV has been hypothesized to behave as “hibernating myocardium,” which may suggest that the RV can be targeted therapeutically in PAH.3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


