Kiran Musunuru, Sekar Kathiresan
Principles of Cardiovascular Genetics
As physicians, we seek to understand the root cause of human disease. Human genetics provides a unique tool for generating new hypotheses about the root causes of disease based on genome-wide searches in the human population that are unlimited by prior assumptions about the underlying pathophysiologic processes. Over the past several decades, application of the principles discussed here has successfully identified the causative genes for a range of cardiovascular diseases. This information has provided explanations to our patients, improved the ability to predict risk for disease, and most importantly, enabled understanding of the pathophysiology as a foundation for designing rational approaches to improving prevention and therapy.1 This chapter reviews the principles of human genetics used to make gene discoveries and to translate these findings to improve patient care. We highlight these principles in the context of a clinical case presentation.
Inherited Basis for the Variation in Risk for Cardiovascular Disease
Patient Case, Part I.
A 44-year-old man (JS) is seen in a cardiologist’s office for a follow-up visit after having suffered an ST-segment elevation myocardial infarction (STEMI) and undergone treatment consisting of primary angioplasty and placement of a drug-eluting stent. His cardiovascular risk factors before STEMI included a fasting low-density lipoprotein cholesterol (LDL-C) level of 235 mg/dL and active cigarette smoking. His body mass index (BMI) is 25 kg/m2, he does not have a history of type 2 diabetes, and he is normotensive. His father died at 45 years of age as a result of myocardial infarction (MI), and his paternal uncle suffered an MI at 49 years of age. He has two brothers, 43 and 39 years old; both are free of clinical cardiovascular disease. The 43-year-old brother (KS) has an elevated LDL-C level (214 mg/dL). The 39-year-old brother (LS) has an LDL-C level of 130 mg/dL and a high-density lipoprotein cholesterol (HDL-C) level of 29 mg/dL. The pedigree of the family is shown in Figure 8-1.
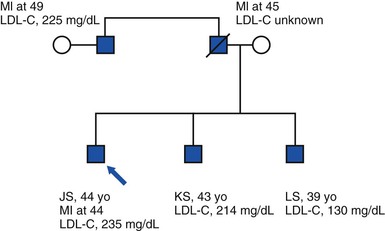
Many cardiovascular diseases cluster within families, and studies of familial aggregation can determine the extent to which inherited DNA sequence variants contribute to these patterns. A family history of premature coronary heart disease (CHD) elevates the risk for CHD in offspring approximately threefold.2 Family history is an important risk factor for almost every cardiovascular disease—including atrial fibrillation, congenital heart disease, and hypertension—but familial clustering of disease can reflect shared environment in addition to shared genetic sequence.
Heritability—the fraction of interindividual variability in risk for disease attributable to additive genetic influences—is a commonly used measure for isolating the role of shared genetic sequence. The remaining variability among individuals results from all other contributors: environmental influences on disease, nonadditive (epistatic) genetic effects (e.g., gene-gene interactions or gene-environment interactions), error in the measurement of relatedness or disease, and random chance. For most clinically important traits (diseases and risk factors), empiric estimates of heritability range from 20% to 80% (see Online Mendelian Inheritance in Man, available at www.ncbi.nlm.nih.gov:80/entrez/query.fcgi?db=OMIM, for comprehensive information).
A Brief Primer on Molecular Biology
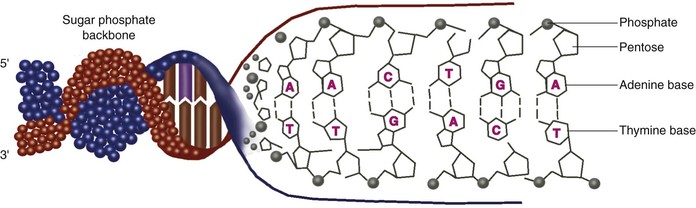
Genes are encoded in DNA, a polymeric molecule with two strands in a configuration known as a double helix. The “code” comprises four different DNA bases—adenine (A), cytosine (C), guanine (G), and thymine (T)—linked together in nonrandom order. The two strands contain redundant information by virtue of complementarity—an adenine on one strand is always paired with a thymine on the other strand, and a cytosine on one strand is always paired with a guanine on the other strand. Thus double-strand DNA can be considered to be a sequence of A-T, T-A, C-G, and G-C base pairs (Fig. 8-2).
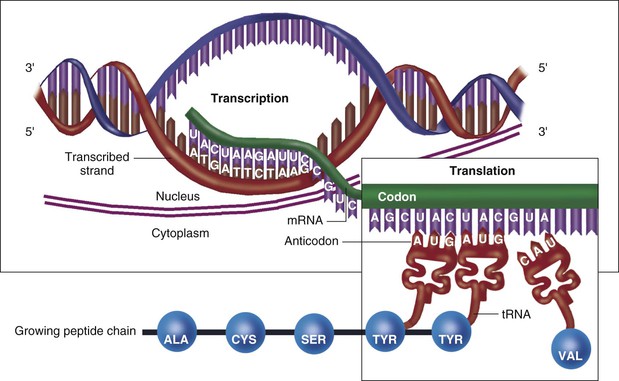
Human DNA is organized into a total of 23 pairs of chromosomes, with each chromosome spanning millions of base pairs. The 46 chromosomes in total make up the genome. Each chromosome has numerous genes, which contain so-called coding DNA, separated by large stretches of noncoding DNA. A process called transcription copies the information in the DNA sequence into single-strand RNA, a polymer that is structurally similar to DNA but uses uracil (U) in place of thymine (T). Subsequently, the process of translation converts the RNA sequence into an amino acid sequence that makes up a protein, which can serve in a variety of roles (structural elements, enzymes, hormones, etc.). Thus genetic information flows from DNA to RNA to protein in what is classically known as the “central dogma” of molecular biology (Fig. 8-3).
One of the consequences of the central dogma is that a change in the DNA sequence in the genome, if it should occur in or near a gene, can result in a change in the protein encoded by the gene, which in turn can have important consequences on the phenotype of an organism. Phenotype refers to any observable characteristic in a human being. Changes in DNA sequence leading to phenotypic changes underlie most of the heritability of diseases that have a genetic component.
Epigenetics pertains to phenotypic changes caused by DNA-level modifications that do not involve the DNA sequence, typically structural modifications either of certain DNA bases or of the proteins (called histones) in which the DNA is packaged. These changes can result in altered levels of RNA being transcribed from the DNA, which in turn results in altered levels of protein. In some cases the epigenetic changes are transmitted from parents to offspring, and thus can represent an additional source of phenotypic heritability.
Modes of Inheritance
The genetic architecture of a disease refers to the number and magnitude of genetic risk factors that exist in each patient and in the population, as well as their frequencies and interactions. Diseases can be due to a single gene (monogenic) in each family or to multiple genes (polygenic). Identifying genetic risk factors is easiest when only a single gene is involved and this gene has a large impact on disease in that family. In cases in which a single gene is necessary and sufficient to cause disease, the condition is termed a mendelian disorder because the disease tracks perfectly with a mutation (in the family) that obeys Mendel’s simple laws of inheritance.
For monogenic disorders, modes of inheritance include autosomal dominant, autosomal recessive, and X-linked. In autosomal dominant disorders, a single defective copy of a gene (either the maternal or paternal copy for every autosomal gene) suffices to cause the phenotype. In autosomal recessive disorders, both copies need to be defective to lead to the phenotype. In X-linked disorders, the defective gene resides on the X chromosome. Given that men have only one X chromosome and women have two X chromosomes, men who carry the defective copy are affected with the disorder, whereas women are unaffected carriers.
Most common cardiovascular diseases, however, do not obey Mendel’s simple laws of inheritance but rather are complex—the result of an interplay between multiple genes and the environment. For these polygenic disorders, variants in more than one gene are needed to cause a disease. Accordingly, in these cases it becomes difficult to understand a disease by studying a single family. A corollary is that each contributing gene variant may have a small phenotypic effect that is not obvious by comparing a few people with and without that variant. For these reasons, elucidating the genetic architecture of a complex disorder is more feasible by studying a large population.
The patient case presented earlier describes both discrete cardiovascular phenotypes (i.e., traits defined by their presence or absence based on a set of criteria) and quantitative phenotypes. MI is a discrete (also called dichotomous) phenotype, whereas blood pressure, LDL-C, HDL-C, and BMI are continuous cardiovascular traits. In the general population, most of these traits display a complex pattern of inheritance.
For many complex traits, however, some subtypes of the disease are monogenic in inheritance. In our patient case, the co-occurrence of high LDL-C, early-onset MI, and a family history of premature MI suggests a specific mendelian disorder, namely, familial hypercholesterolemia (FH).3 In FH, the extremely high LDL-C level and MI result from defects in the LDL receptor gene. Severely high LDL-C and early MI can also be caused by defects in other genes, including proprotein convertase subtilisin/kexin type 9 (PCSK9) and apolipoprotein B (APOB). Other examples of monogenic subtypes of complex traits include extremely high or low blood pressure caused by rare mutations in genes involved in renal salt handling; extremely low LDL-C as a result of mutations in APOB, PCSK9, or ANGPTL3; and extreme obesity caused by mutations in MC4R.
Approaches to Discovering the Inherited Basis for Cardiovascular Disease
Human Genetic Variation
The human genome contains about 6 billion base pairs across the 46 chromosomes. Approximately 1% of the genomic DNA is coding DNA, which comprises an estimated 20,000 genes.4 Although most of the DNA in the genome is shared among all human beings, variations in the DNA sequence—occurring in both coding DNA and noncoding DNA—distinguish individuals from one another. These DNA sequence variants partly account for why a disease is more or less likely to develop in some individuals or why some respond more favorably or more adversely to a medication (see also Chapters 7 and 9).
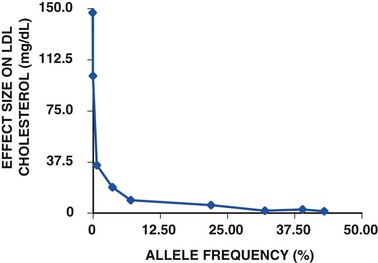
As alluded to earlier, some DNA sequence variants have large phenotypic effects—meaning that they can cause disease singlehandedly. These DNA sequence variants tend to be rare (and sometimes unique to a single person or family) because natural selection weeds them out of a population. Classically, they cause monogenic disorders. Other DNA sequence variants commonly occur in a population and tend to have smaller phenotypic effects. Typically it is these variants, in combination, that cause polygenic disorders. Because of natural selection, in general there is an inverse relationship between the frequency of a DNA sequence variant and the phenotypic effect conferred by that variant. For example, such a relationship is observed for gene variants that affect LDL-C in the population (Fig. 8-4).5–8
Coding sequence variants potentially disrupt the function of genes and their protein products (Fig. 8-5).9 Some coding variants do not affect the amino acid sequence of a protein; these are known as synonymous variants and do not usually have any phenotypic consequences. Other coding variants can cause a variety of alterations in a protein—substitution of a single amino acid in a protein with a different amino acid (missense), premature truncation of a protein (nonsense), scrambling of the amino acid sequence past the site of the variant (frameshift), or insertion or deletion of amino acids. Any of these so-called nonsynonymous variants can have phenotypic effects ranging from negligible to profound, although nonsense and frameshift variants tend to be more deleterious than missense variants to protein function. Finally, sequence variants at splice sites (the first and second bases after the end of each exon and before the beginning of each exon) can lead to a severely disrupted protein product missing an entire exon.
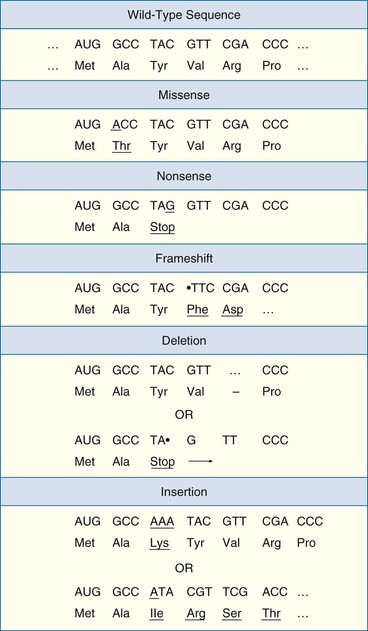
Noncoding variants, although they do not directly affect the amino acid sequences of proteins, can cause phenotypic changes in other ways. For example, a noncoding variant near a gene might affect transcription of the gene and result in an increased amount of RNA being produced from a gene, and consequently an increased amount of the protein product.10 Noncoding variants can affect the processing of RNA in several other ways.
In addition to genes, the genome harbors a number of expressed RNA molecules that do not code for protein; such RNA includes microRNA and large intergenic noncoding RNA (lincRNA). Both these categories of noncoding RNA have been demonstrated to interact with and modulate the activity of coding RNA, thereby regulating protein levels. For example, a given microRNA might physically bind to complementary sequences in a large number of coding RNA molecules and result in either suppression of RNA translation into proteins or degradation of the RNA. A noncoding variant that falls in the midst of a microRNA might impair (or enhance) its ability to interact with specific coding RNA and result in phenotypic changes.
DNA sequence variants, also known as polymorphisms (derived from Greek words meaning “multiple forms”), consist of three major classes. Single-nucleotide polymorphisms (SNPs) involve the alteration of a single DNA base pair in the genome. They are the most common and best cataloged of the DNA variants, with tens of millions having been identified to date across all human populations. Variable number tandem repeats (VNTRs) involve a variable number of repeats of a short DNA sequence at a genomic location; the number of repeats ranges from very few to thousands. Copy number variants (CNVs) involve a variable number of repeats of a long DNA sequence (more than 1000 base pairs), typically ranging from zero to one or a few repeats. An indel (an abbreviation of insertion/deletion) is a type of DNA variant in which a sequence is either present (insertion) or absent (deletion); it could be either a special type of a VNTR or a special type of a CNV, depending on the size of the involved sequence.
Characterizing Human Genetic Variation: Genotyping and Sequencing
In most cases a person has two copies of each DNA sequence because of the presence of paired chromosomes (the exceptions are DNA sequences on the X or Y chromosome in men, because these two chromosomes are entirely different). The two copies are known as alleles. For a DNA variant, the genotype is the identity of the two alleles at the site of the variant. The two alleles may be identical, in which case the person is said to be homozygous for the allele. If the two alleles are different, the person is heterozygous at the DNA variant. A haplotype is a series of genotypes at nearby sites of DNA variants. Because the haplotype is located on a single region of the chromosome, it tends to remain linked together as it passes from parents to offspring.
For polymorphisms that are primarily present in just two forms (typical of SNPs, i.e., one DNA base versus another DNA base, but not for VNTRs, which are usually found in at least a few forms, i.e., different numbers of repeats), the allele found more commonly in a given population is termed the major allele, with the less common allele being the minor allele. Common variants are so defined by virtue of the frequency of the minor allele being greater than 5% in the population. Low-frequency variants have a minor allele frequency of between 0.5% and 5%; rare variants have less than a 0.5% frequency. Rare variants are typically referred to as mutations. In some cases, mutations are so rare that they are found only in one individual or in one family.
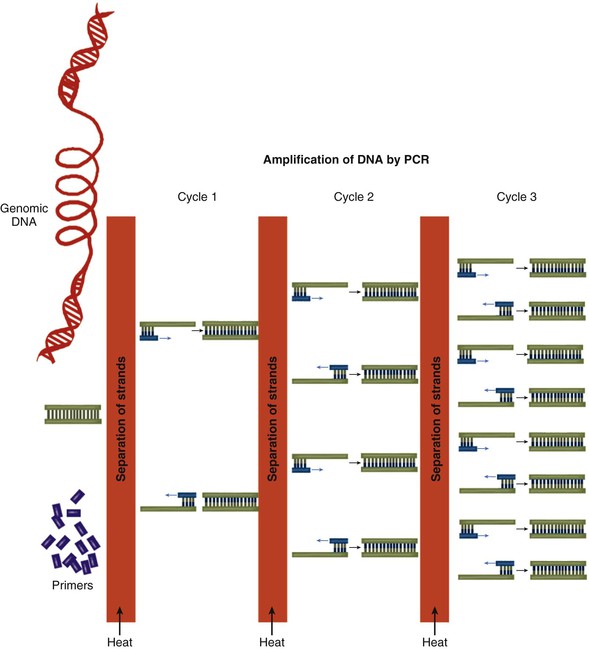
Two types of methods can be used to determine genotypes at the sites of DNA variants. In the first type, a genotyping technology directly ascertains the genotype at a single location in the genome. In the second type, polymerase chain reaction (PCR) is used to amplify the region of DNA immediately surrounding the site of the DNA variant (Fig. 8-6). The PCR product is subjected to DNA sequencing, which indirectly determines the genotype. The first type is generally cheaper—indeed, fabricated “chips” can directly genotype millions of DNA variants at a time—but requires optimization beforehand. Thus direct genotyping is most useful for common and low-frequency variants that have already been cataloged. The second type is more expensive and can be used only at one location at a time, but it can be flexibly adapted to any location in the genome. This approach can be used to discover previously uncataloged rare DNA sequence variants.
In recent years, a third type of method has been devised to characterize a person’s genetic variation. This method entails the use of any of a group of techniques known as next-generation DNA sequencing.11 Although the operational details differ, these techniques share the ability to sequence billions of DNA base pairs at a time within a reasonable time frame and at a reasonable cost. The techniques have been applied successfully to efficiently sequence the entirety of a patient’s coding DNA, known as the exome, which accounts for about 1% of the genome.12,13 More recently, sequencing the entirety of a patient’s genome for a few thousand U.S. dollars within 24 hours has become feasible, with the highly publicized “thousand-dollar genome” expected to emerge very soon.
Although performing DNA sequencing remains more expensive than direct genotyping, the decreasing cost of whole-genome sequencing will soon enable it to be performed in large cohorts of people. The advantage of whole-genome sequencing is that it determines genotypes at the locations of all known DNA sequence variants in a single experiment and, at the same time, identifies previously unknown DNA variants that are unique to the individual.
Study Designs to Correlate Genotype with Phenotype
Approaches to correlate genotype with phenotype are highlighted in Figure 8-7. The x axis shows the frequency of the allele in the population, from rare to common; the y
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


