Chapter 14 Primary Pulmonary Hypertension
Pulmonary arterial pressure was measured in 1852,1 but a century elapsed before cardiac catheterization provided the means for studying the physiology of the lesser circulation. Elevated pulmonary arterial pressure—pulmonary hypertension—results from disorders of the pulmonary vascular bed, the pulmonary parenchyma, the respiratory system, and the pulmonary venous bed and from chronic thromboembolic disease (Box 14-1).2 This chapter focuses on the pulmonary vascular bed, specifically on primary (idiopathic) pulmonary hypertension, a disorder that originates in the terminal muscular pulmonary arteries and arterioles.3,4
The earliest description of idiopathic pulmonary hypertension was a report in 1891 of the cardiac pathology in a patient with pulmonary artery sclerosis and right ventricular hypertrophy of unknown cause.5 Primary pulmonary hypertension, a term coined by Dresdale in 1951, referred to an idiopathic disorder residing in the terminal pulmonary arteries and arterioles. The prevalence rate of primary pulmonary hypertension is estimated at one or two cases per million in the general population.3 Shortly after Dresdale’s report, Wood6 characterized the clinical manifestations of primary pulmonary hypertension. Two decades later, the World Health Organization (WHO) proposed a classification based on three histopathologic patterns: plexogenic pulmonary arteriopathy, microthrombotic pulmonary arteriopathy, and pulmonary venoocclusive disease.7 In 1975, WHO defined primary pulmonary hypertension as a disorder with no identifiable cause in which resting mean pulmonary artery pressure in adults at sea level is above 25 mm Hg and above 30 mm Hg with exercise. Pulmonary vascular resistance does not exceed three Wood units, and pulmonary capillary wedge pressure is normal.3,4,7 In 1981, the National Heart, Lung, and Blood Institute established a Registry to collect information on primary pulmonary hypertension in accordance with a uniform database.8 Enlightening histopathologic studies abound.8–13
The pulmonary vascular bed consists of an elaborately branched system of arteries, arterioles, capillaries, venules, and veins that accommodate the cardiac output at low pressures. Approximately 17 gradations of arterial vascular channels separate the main pulmonary artery from the pulmonary arterioles. In the fetus, the pulmonary arteriole is structurally analogous to the systemic arteriole and is therefore equipped to meet the full force of systemic pressure the moment the neonatal lungs expand and pulmonary blood flow commences. The thick-walled neonatal pulmonary arterioles rapidly involute, and the pulmonary vascular bed remodels, establishing the low-resistance lesser circulation. Remodeling continues for 1 or 2 months as the lungs adapt to extrauterine life and then proceeds slowly throughout childhood as the pulmonary vascular bed matures.14
The major airways and the major pulmonary arterial branches are formed by 16 weeks of gestation. An understanding of the density per unit area of small peripheral pulmonary arteries relative to alveolar density is necessary to understand normal and abnormal fetal and postnatal pulmonary arterial development.15 The number of peripheral acinar units increases in proportion to the number of intraacinar arteries until lung growth is completed at 8 to 10 years of age. Two thirds of the normal number of intraacinar arteries are formed by 18 months, and most of the remainder are formed by 5 years of age.15 Also important is the normal age-related distal extension of medial smooth muscle.15 At birth, the vascular channels beyond the terminal bronchioles are devoid of medial smooth muscle, but within 2 to 3 years, medial smooth muscle extends to the junction of respiratory bronchioles and alveolar ducts and, by the mid-teens, extends among alveoli into precapillary vessels.
The histologic findings in primary pulmonary hypertension (see previous discussion) include luminal hyperplasia, medial hypertrophy, adventitial proliferation and fibrosis, occlusion of small arteries, in situ thrombosis, and infiltration of inflammatory and progenitor cells.4 In 1958, Heath and Edwards16 published elegant descriptions of the histology of pulmonary vascular disease together with a comprehensive classification.17 The earliest abnormality was muscular thickening of small pulmonary arteries from medial hyperplasia, followed by intimal thickening (hyperplasia) that ranged from minimal to virtual luminal occlusion. The end stage was the angioproliferative plexiform lesion that corresponds to one of the three major histopathologic types of primary pulmonary hypertension in the WHO classification.7 Originally, plexiform lesions were thought to be unique to primary pulmonary hypertension,4 but now the belief is that plexogenic pulmonary arteriopathy is a nonspecific response of the pulmonary vasculature to hemodynamic injury.3 The progression of lesion severity from medial hypertrophy to plexiform arteriopathy and the functional relevance of plexiform lesions remain uncertain.4
Plexogenic pulmonary arteriopathy resides in small muscular arteries 100 to 200 μm in diameter and is a complex process that involves intimal cell proliferation, migration of medial smooth muscle cells, recruitment of inflammatory cells, and deposition of extracellular matrix proteins.8,12,18 Intimal proliferation is disproportionate to medial hypertrophy and culminates in complete luminal obliteration. Plexiform lesions are found in 70% to 85% of patients with primary pulmonary hypertension in whom lung tissue is available.3,12
Microthrombotic pulmonary arteriopathy occurs in 20% to 50% of cases of primary pulmonary hypertension12 and is represented by recanalized in situ microthrombi that consist of fibrous webs with eccentric intimal fibrosis together with medial hypertrophy but without plexiform lesions.8,12,18
Pulmonary venoocclusive disease is found in less than 7% of cases of primary pulmonary hypertension12 and is characterized by fibrosis and intimal proliferation of intrapulmonary venules, culminating in complete obliteration of venous channels that tend to recanalize.8,12,18,19 Studies with use of light and electron microscopy have confirmed and extended these observations of pulmonary vascular pathophysiology (Box 14-2).20 The clinical manifestations of primary pulmonary hypertension do not distinguish among the histopathologic types of pulmonary vascular disease. Early vasoreactivity ultimately becomes fixed,11 but once the disorder is established, it rarely regresses.12,16,17,21,22
Advances in pulmonary vascular biology continue to shed light on the pathogenesis of primary pulmonary hypertension.23–25 Endothelial abnormalities have been identified (vasoconstrictor endothlin-1 homeostasis),26–28 together with defects in voltage-gated potassium channels,4 abnormalities in metalloproteinase and elastase activity,4,29 transforming growth factor beta,30,31 and bone morphogenetic protein.32 Mediators of inflammation can cause pulmonary vasoconstriction, but the role of inflammation in chronic pulmonary hypertension is less clear. Genetic mechanisms are generally accepted to play a pathogenetic role in familial and sporadic cases of primary pulmonary hypertension (see section The History).4,32–36 Persistent fetal circulation or persistent pulmonary hypertension of the newborn, a disorder of unknown etiology that was described in 1969,37 is characterized by muscularization of small pulmonary arteries in the fetal lung, high neonatal pulmonary vascular resistance that fails to regress, and persistent right-to-left shunts through the ductus arteriosus and foramen ovale.37–40 A relationship between persistent fetal circulation and primary pulmonary hypertension is tenuous.41Portal hypertension can be associated with pulmonary hypertension presumably because the pulmonary vascular bed is exposed to vasoreactive substances normally metabolized by the liver.42,43 An association between human immunodeficiency infection and hypertensive pulmonary arteriopathy has been reported.44Alveolar hypoventilation and sleep apnea caused by chronic upper airway obstruction of hypertrophied tonsils and adenoids in children result in an increase in pulmonary vascular resistance (see Box 14-1).45–48 High-altitude pulmonary hypertension clinically and histologically resembles primary pulmonary hypertension, which is more common at high altitudes and which is aggravated by the low partial pressure of oxygen (see section The History).49–51
The physiologic consequences of primary pulmonary hypertension are direct reflections of the increased resistance to blood flow through the lesser circulation. The pressure in the pulmonary arteries can exceed systemic pressure (Figure 14-1). Pulmonary vasoreactivity can be present initially but diminishes and is ultimately lost. Resting pulmonary blood flow and cardiac output fall and either fail to increase during exercise or fall still further.52 Asymptomatic gene carriers for primary pulmonary hypertension have been identified with abnormal responses to exercise.53
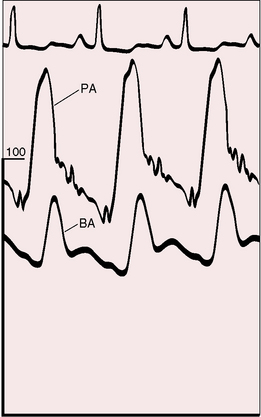
Forceful right atrial contraction generates a large A wave and an increase in right ventricular end-diastolic segment length. Right ventricular end-diastolic pressure rises in response, right atrial mean pressure rises, and a right-to-left shunt is established through a stretched patent foramen ovale. Tricuspid regurgitation increases mean right atrial pressure still further.54 Systemic venous pressure can be sufficiently elevated to cause venous stasis retinopathy.55,56
The atrial natriuretic peptide system is activated in primary pulmonary hypertension in response to increased right atrial pressure.57 The polypeptide hormone is secreted by atrial myocytes; is involved in the homeostatic control of body water, sodium, and potassium; and correlates significantly with right ventricular preload and afterload.57Brain natriuretic peptide levels increase in proportion to right ventricular dysfunction.58
A mild albeit significant reduction in total lung capacity occurs in females with primary pulmonary hypertension, and a reduction in forced vital capacity occurs in both males and females.8 Hypoxemia and hypocapnia are almost invariable because of a ventilation-perfusion mismatch, not because of a right-to-left shunt across a patent foramen ovale.8 The diffusing capacity for carbon monoxide is significantly less than predicted.8
The ventricular septum encroaches on the left ventricular cavity, sometimes appreciably.59 End-systolic and early diastolic deformations of the left ventricular cavity result in impaired filling in early diastole and in redistribution of filling in late diastole.59–61 Early diastolic filling of the right ventricle is also encroached upon because high pulmonary vascular resistance prolongs the duration of right ventricular systole and prolongs isovolumetric relaxation.61
The time course of pulmonary hypertension can be established from the histology of the pulmonary trunk compared with the histology of the aortic root.17,62,63 When pulmonary hypertension begins at birth, the histology of the pulmonary trunk and the histology of the aortic root are virtually identical (Figures 14-2C and 14-2D). When pulmonary hypertension begins after birth, the histology of the pulmonary trunk and aortic root differ significantly (Figures 14-2A and 14-2B). Primary pulmonary hypertension is rarely present at birth,17,62,63 but when that is the case, it serves as a model of isolated pure pulmonary hypertension with which pulmonary hypertension with congenital heart disease can be compared.

History
The frequency of primary pulmonary hypertension in young females was cited as early as 1927,64 with a female:male ratio subsequently reported as high as 3:1.9 In the National Institutes of Health (NIH) registry, however, the gender incidence rate in children was equal; and in adults, the female:male ratio was 1.7:1, with a relatively constant incidence rate decade by decade.8 Oral contraceptives are a consideration in light of evidence that birth control pills can aggravate if not initiate pulmonary vascular disease.65
Variations in gender incidence may in part reflect differences among the three histopathologic subgroups. Plexogenic pulmonary arteriopathy, the most frequent of the three histopathologic subgroups of primary pulmonary hypertension,8,18 has a female:male ratio of 3:1. In the less common microthrombotic pulmonary hypertension, the female:male ratio is approximately equal,18 and in the venoocclusive subgroup, which accounts for less than 7% of cases, a slight male predominance is seen.12
Familial primary pulmonary hypertension is well recognized (see Figure 14-12A) and occasionally recurs in more than one generation.66–71 In the NIH registry, 6% of patients with primary pulmonary hypertension had a first-degree relative with primary pulmonary hypertension.3 Inheritance is autosomal dominant with incomplete penetrance.53 The genetic basis has been assigned to mutations in the bone morphogenetic protein receptor.72–74
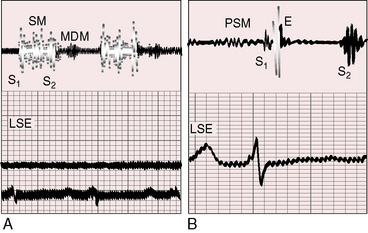
Figure 14-12 A, Phonocardiogram from a 20-year-old man with primary pulmonary hypertension. His younger sister also had primary pulmonary hypertension. The tracing at the lower left sternal edge (LSE) shows a holosystolic murmur of tricuspid regurgitation and a short middiastolic murmur (MDM). B, Phonocardiogram from the 29-year-old woman referred to in Figure 14-5. A short presystolic murmur (PSM) and a prominent pulmonary ejection sound (E) are shown at the lower left sternal edge. (S2 = second heart sound.)
Mean patient age in three large studies of primary pulmonary hypertension was 21 to 30 years, with a range of 2 to 56 years.75 In the NIH registry, only 8% of patients reached the seventh decade,8 although primary pulmonary hypertension of long duration has been reported. Regression has been suspected, albeit rarely.21,22 Short-term survival is related to the severity of endothelial dysfunction.27 Median survival after diagnosis is approximately 2.8 years,3,76 with right ventricular failure the most common cause of death.76–78 Longevity and morbidity are significantly influenced by lifestyle. Abrupt, strenuous, or isometric exercise should be avoided, and isotonic exercise should cease at the very onset of dyspnea, light-headedness, dizziness, or chest pain. Exposure to high altitude is an avoidable risk, and certain constraints are appropriate for air travel. A commercial jetliner flying at 33,000 to 36,000 ft has a cabin atmosphere equivalent to approximately 6000 to 8000 ft, which is comparable with breathing 15% oxygen at sea level.17,79 Airline passengers experience a significant decrease in arterial PO2, but only a mild decrease in arterial oxygen saturation.79 Low humidity causes dehydration and an undesirable fall in systemic blood pressure. Just as important are the risks incurred by non–flight-related stress and travel fatigue. Rushing at the last minute, carrying heavy baggage, and transferring from one terminal to another at large airports are recognized risks.
The most common symptoms associated with primary pulmonary hypertension are effort dyspnea, fatigue, light-headedness, and chest pain. An increase in ventilatory response to exercise—hyperventilatory dyspnea—is attributed chiefly to a ventilation/perfusion mismatch.12,52
Muscle fatigue is attributed to a reduction in the rate of aerobic regeneration of adenosine triphosphate (ATP).52 Hypothyroidism is surprisingly prevalent in primary pulmonary hypertension80 and should be considered in patients with inappropriate fatigue. Stress-related or exercise-related light-headedness, giddiness, dizziness, or faintness reflects an inability to achieve sufficient cardiac output and systemic blood pressure to maintain cerebral blood flow. The chest pain of myocardial ischemic originates in the hypertrophied hypoperfused right ventricle.81 Histologic evidence of right ventricular infarction occurs in patients with primary pulmonary hypertension and normal coronary arteries. Myocardial ischemia has also been attributed to compression of the left main coronary artery by a dilated hypertensive pulmonary trunk.82,83 Syncope, which is usually provoked by effort or excitement, is ominous and heralds sudden death.8,12,84 Sudden death can also follow relatively innocuous stress, such as bone marrow aspiration and cardiac catheterization, especially in children.77,85 Surgery, anesthesia, and even sedatives are poorly tolerated. Symptoms may first appear during pregnancy, which warrants special emphasis.86–90 Fixed pulmonary vascular resistance blunts or precludes adaptive responses to the hemodynamic fluctuations of labor, delivery, and the puerperium. Maternal mortality rate is as high as 50%.88 A hypercoagulable state in the third trimester reinforces the propensity for in situ thromboses in small terminal pulmonary arteries (see previous discussion).
Hemoptysis is a feature of Eisenmenger’s syndrome (see Chapter 17) but not primary pulmonary hypertension. Raynaud’s phenomenon is occasionally associated with primary pulmonary hypertension and calls attention to autoimmunity (see previous discussion).24 Hoarseness, Ortner’s syndrome, results from compression of the left recurrent laryngeal nerve by a dilated hypertensive pulmonary trunk.12,69 Patients are sometimes unpleasantly aware of visible neck pulsations caused by giant jugular A waves (Figure 14-3; see previous discussion).
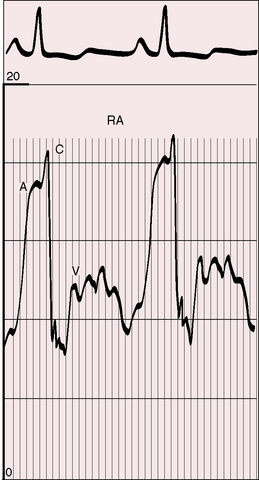
Figure 14-3 Right atrial pressure pulse from the 18-year-old man referred to in Figure 14-1 shows A waves of 14 to 16 mm Hg.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


