Abstract
Primary ciliary dyskinesia (PCD) is one of an expanding collection of disorders collectively known as ciliopathies. A rare, inherited disease of the motile cilia, PCD is clinically characterized by chronic upper and lower airway disease, left-right laterality defects, and infertility caused by ciliary (or flagellar) dysfunction. For four decades, the diagnosis of PCD has been based on the presence of ultrastructural defects in the ciliary axoneme using transmission electron microscopy analysis, but newer diagnostic tests have recently emerged, including nasal nitric oxide measurement, high-speed videomicroscopy with ciliary beat pattern analysis, and immunofluorescence imaging for specific axonemal proteins. Genetic testing has also become an increasingly useful diagnostic alternative. Indeed, a growing number of PCD-associated genes have been identified, which encode proteins essential for ciliary assembly, structure, and function, which has advanced our understanding of the disease. In this chapter, we will review the genetics and pathophysiology of PCD, describe emerging diagnostic tests, and outline current therapies for this rare lung disease.
Keywords
bronchiectasis, cilia, ciliopathies, dynein, heterotaxy, microtubules, nasal nitric oxide, nexin, radial spoke, situs ambiguous, situs inversus totalis
Epidemiology
Eighty years ago, Kartagener described a clinical syndrome characterized by the triad of situs inversus totalis, chronic sinusitis, and bronchiectasis that later was defined as Kartagener syndrome. Alfzelius and colleagues later found that individuals with Kartagener syndrome, as well as others with chronic sinusitis and bronchiectasis, have defects in the ultrastructural organization of motor cilia. Initially, the term “immotile cilia syndrome” was used to describe this disorder, but subsequent studies showed that cilia are often motile but their beat uncoordinated and ineffective. Thus the name was changed to “primary ciliary dyskinesia” (PCD) to more appropriately describe the spectrum of ciliary dysfunction and to distinguish “primary” or genetic ciliary defects from “secondary” or acquired defects associated with epithelial injury.
PCD is an inherited disorder characterized by impaired motor ciliary function leading to diverse clinical manifestations. The frequency of PCD is approximately between 1 in 12,000 and 20,000 live births, based on the prevalence of situs inversus totalis and bronchiectasis in population surveys, but these values likely underestimate its incidence in the general population. Although PCD is considered a rare lung disease, its prevalence in children with repeated respiratory infections has been estimated to be as high as 5%.
Our understanding of the epidemiology, genetics, pathophysiology, and clinical manifestations of PCD has rapidly advanced over the four decades since the disease was linked to ultrastructural defects of the ciliary axoneme. Advances in cilia genetics and biology have provided new insights into genotype-phenotype relationships and may yield potential therapeutic targets to restore ciliary structure and function.
Etiology
PCD is a heterogeneous genetic disorder, usually autosomal recessive, of motile ciliary structure function and biogenesis. A wide spectrum of clinical features may be seen, reflecting the numerous organs where cilia are important for maintaining health. The large airways and contiguous structures, such as the nasopharynx, paranasal sinuses, and middle ear, are lined by a ciliated, pseudostratified columnar epithelium that is important for mucociliary clearance. The cells on the lumenal surface of this epithelium are predominantly ciliated cells with interspersed goblet cells. Other epithelia containing motile ciliated cells are found in the ependyma of the brain and the fallopian tubes. Spermatozoa flagella have a core structure similar to that of cilia with the same fundamental motility characteristics.
Normal Motile Ciliary Structure and Function
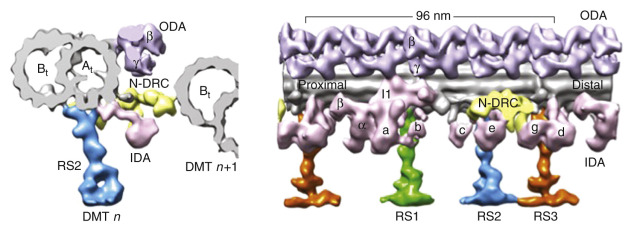
Mature respiratory ciliated cells contain approximately 200 cilia of uniform size, with an average length of 6 µm. Each normal cilium contains an array of longitudinal microtubules, consisting of nine doublets arranged in an outer circle around a central pair ( Fig. 71.1 ). The microtubules are anchored by a basal body in the apical cytoplasm of the cell. Several different proteins contribute to ciliary structure and function. Tubulin, a dimeric molecule with α- and β-subunits, forms long microtubules that extend the entire length of the cilium. These microtubules are arranged in a distinct 9+2 pattern, with 9 microtubule doublets arranged in a circular array around a central pair of microtubules. The protein nexin links the outer microtubular doublets, creating a circumferential network, and radial spokes connect the outer microtubular doublets with a central sheath of protein that surrounds the central tubules. Dynein is attached to the microtubules as distinct inner and outer “arms,” recognizable on electron micrographs of axonemal cross-sections, and is thought to participate in the provision of energy for microtubule sliding through adenosine triphosphatase (ATPase) activity. Inner and outer dynein arms are attached to each microtubule doublet at precise, repeated intervals—24 nm for outer dynein arms and 96 nm for inner dynein arms. The outer dynein arms are longer and form a hook, whereas the inner dynein arms are straight and linked to radial spokes. Each dynein arm is a multimer of two or three heavy chains (400–500 kd), two or four intermediate chains (45–110 kd), and at least eight light chains (8–55 kd), with each chain protein encoded by a distinct gene. The dynein heavy chains contain the ATPase that provides the energy for ciliary movement.

Dynein arms extend from the A tubule and interact with the B tubule of the neighboring outer pair, and the force generated translates to a sliding motion of two neighboring tubules, while the inner dynein arms are central for controlling the rhythmic motion of cilia. The inner dynein arm is also part of a complex referred to as the dynein regulatory complex, a key regulator of motor activity. Located within the nexin link, an elastic element that limits sliding between microtubules. The dynein regulatory complex consists of several closely associated proteins that coordinate the activity of the multiple dyneins. The radial spokes also regulate dynein arm activity, sending signals from the central apparatus to the dynein arms. All these structures work in a coordinated fashion to produce a synchronized ciliary beat and maintain alignment of the doublet microtubules ( Fig. 71.2 ).
Ciliary motion takes place within an aqueous layer of airway surface liquids and is divided into two phases: an effective stroke phase that sweeps forward; and a recovery phase during which the cilia bend “backward” and extend into the starting position for the stroke phase. Typically, the tips of cilia contact the overlying mucus during the stroke phase to propel mucus forward, but during the recovery phase, cilia lose contact with the mucus. Normal human cilia bend in a rhythmic, wavelike motion within a single plane. The normal beat frequency of human cilia ranges between 8 and 14 Hertz, which can vary with testing conditions. Cila beat quicker in the proximal airways than in more distal airways and faster in young children (13 beats/s) than in adults (11.5 beats/s). In healthy epithelium, cilia are aligned spatially with parallel orientation of the central pair of tubules in adjacent cilia; therefore ciliary motility is maintained in the same general orientation along the length of airways. Normal mucociliary transport rates may be as rapid as 20–30 mm/min. Although the intracellular and intercellular regulation of ciliary actions is complex and not fully understood, several signaling mechanisms including intracellular calcium, cyclic adenosine monophosphate (cAMP), extracellular adenosine triphosphate (ATP), and airway nitric oxide (NO) play key roles in regulating ciliary beat frequency. Ciliary motion is synchronized along the respiratory epithelium by calcium signaling through gap junctions. Motile cilia also respond to external stimuli, such as infectious agents and airborne pollutants, suggesting that motile cilia can sense their environment like primary cilia.
There are other types of cilia in the body ( Fig. 71.3 ). Nodal cilia exist transiently in the ventral node of the gastrula during embryonic development. The ultrastructure of embryonic nodal cilia has many features of epithelial motile cilia, including the circular array of 9 microtubule doublets with inner and outer dynein arms; however, there is no central pair of microtubules, hence a 9+0 array. This structure allows the organelle to rotate in a whirling motion that generates unidirectional flow of fluid critical for directing left-right asymmetry in the developing embryo. The node includes two populations of cilia: motile and sensory —the motile nodal cilia generate leftward flow of extracellular fluid across the surface that is detected by peripheral sensory cilia, which activates a cascade of transcription and growth factors to establish body sidedness. In the absence of leftward flow, left-right laterality becomes a random event.

Primary (sensory) cilia are solitary structures present on the surface of many nondividing cells, as well as epithelia-lined sensory organs, biliary ductules, renal tubules, chondrocytes, and astrocytes. These structures were long considered vestigial remnants of no physiologic significance but are now recognized as vital signaling organelles that sense the extracellular environment. Primary cilia serve as chemoreceptors, mechanoreceptors, osmoreceptors, and, in specialized cases, changes in light, temperature, and gravity. Development, growth, and repair functions are also mediated by primary cilia through surface receptors, and genetic defects are associated with clinically diverse pediatric conditions, collectively known as ciliopathies. Primary cilia have a 9+0 structure like nodal cilia but do not have dynein arms and consequently are not motile. Otherwise, primary cilia and motile cilia share many proteins and structures, and there are several lines of evidence that suggest motile cilia have sensory and signaling functions. Defects in the assembly and function of primary cilia have been linked to a wide variety of disorders termed primary ciliopathies , including polycystic kidney disease, Bardet-Biedl syndrome, Joubert syndrome, and retinitis pigmintosa ( Fig. 71.4 ). New insights into the molecular basis for primary ciliopathies has raised interest in potential overlap between sensory and motor ciliopathies.

Pathology and Pathogenesis
Genetics
PCD has been reported in diverse ethnic groups without apparent racial or gender predilection. In most families, PCD appears to be transmitted by an autosomal-recessive pattern of inheritance; however, rare instances of autosomal-dominant or X-linked inheritance patterns have been reported. Parents of affected children are normal and have no evidence of impaired ciliary structure or function.
Genetic heterogeneity of PCD is suggested by its various ultrastructural phenotypes. The conservation of cilia and flagellar structures along the phylogenetic tree, from algae (Chlamydomonas reinhardtii) to flies (Drosophila melanogaster) to fish (Danio rerio) to mammals, has provided insights into the genetics of the human cilium. For instance, C. reinhardtii, a biflagellated single-cell organism, has been a widely used as a powerful model to study motile cilia. The entire genome for Chlamydomonas has been sequenced, and genetic analyses in dysmotile mutant strains of Chlamydomonas have identified numerous orthologous human genes that are mutated in PCD. Indeed, most of the mammalian genes linked to PCD have Chlamydomonas orthologs. The identification of PCD-associated genes has relied on a combination of experimental models, creation of cilia-related transcriptomes and proteomes, and powerful sequencing techniques of candidate genes. Massive parallel sequencing has permitted rapid identification of new mutations in PCD subjects without previous knowledge of candidate genes.
Initial genetic studies focused on genes encoding dynein arm proteins but more recently expanded to include other proteins in ciliated cells. Through a collaborative, international research effort, great progress has been made in identification of PCD genes. To date, more than 35 genes have been liked to PCD ( Table 71.1 ), and approximately 70% of all patients tested have biallelic mutations of these genes. As gene discovery continues, that number will continue to rise. Many of the gene mutations have been linked to specific ultrastructural defects and ciliary dysmotility, including genes that encode outer dynein arm components: DNAH5 (MIM 603335 ), DNAI1 (MIM 604366 ), DNAL1 (MIM 610062 ), DNAI2 (MIM 605483 ), TXNDC3 (MIM 607421 ), and CCDC114 (MIM 615038 ); inner dynein arm, dynein regulatory complex, and nexin components: CCDC39 (MIM 613798 ) and CCDC40 (MIM 613799 ); or the radial spokes and central apparatus: RSPH9 (MIM 612648 ), RSPH4A (MIM 612647 ), and HYDIN (MIM 610812 ). More recently, mutations in genes coding for several cytoplasmic proteins not integral to the cilia axoneme have been reported. These genes encode proteins that presumably have roles in cilia assembly or protein transport, including: HEATR2 (MIM 614864 ), DNAAF1 (MIM 613190 ), DNAAF2 (MIM 612517 ), DNAAF3 (MIM 614566 ), CCDC103 (MIM 614677 ), LRRC6 (MIM 614930 ), DYX1C1 (MIM 608706 ), CCDC114 (MIM 615038 ), SPAG1 (MIM 603395 ), and ZYMND10 (607070) ( Fig. 71.5 ).
| Human Gene | Encoded Protein | Protein Location | Ultrastructural Defect |
|---|---|---|---|
| DNAH5 | Dynein heavy chain 5 | ODA-HC | Absent or truncated ODA |
| DNAH11 | Dynein heavy chain 11 | ODA-HC | Normal ultrastructure |
| DNAI1 | Dynein Intermediate chain 1 | ODA IC | Absent or truncated ODA |
| DNAI2 | Dynein intermediate chain 2 | ODA IC | Absent or truncated ODA |
| DNAL1 | Dynein light chain 1 | ODA-LC | Absent or truncated ODA |
| TXNDC3 | Thioredoxin domain–containing protein 3 | ODA LC/IC | Absent or truncated ODA |
| CCDC103 | Coiled-coil domain–containing protein 103 | ODA docking | Absent or truncated ODA |
| CCDC114 | Coiled-coil domain–containing protein 114 | ODA docking | Absent or truncated ODA |
| DNAAF1 | Dynein (axonemal) assembly factor 1 | Cytoplasm | Absent ODA and IDA |
| DNAAF2 | Dynein (axonemal) assembly factor 2 | Cytoplasm | Absent ODA and IDA |
| DNAAF3 | Dynein (axonemal) assembly factor 3 | Cytoplasm | Absent ODA and IDA |
| HEATR2 | HEAT repeat–containing protein 2 | Cytoplasm | Absent ODA and IDA |
| LRRC6 | Leucine-rich repeat–containing protein 6 | Cytoplasm | Absent ODA and IDA |
| SPAG1 | Sperm-associated antigen-1 | Cytoplasm | Absent ODA and IDA |
| ZMYND10 | Zinc finger, MYND-type containing 10 | Cytoplasm | Absent ODA and IDA |
| DYX1C1 | Dyslexia susceptibility 1 candidate 1 | Cytoplasm | Absent ODA and IDA |
| CCNO | Cyclin O | Cytoplasm | Reduced numbers of cilia |
| MICDAS | Multicilin | Cytoplasm | Reduced numbers of cilia |
| CCDC39 | Coiled-coil domain–containing protein 39 | DRC | Absent IDA with axonemal disorganization in some cilia |
| CCDC40 | Coiled-coil domain–containing protein 40 | DRC | Absent IDA with axonemal disorganization in some cilia |
| CCDC65 | Coiled-coil domain–containing protein 65 | DRC | Normal ultrastructure |
| CCDC164 | Coiled-coil domain–containing protein 164 | DRC | Normal ultrastructure |
| RSPH1 | Radial spoke head protein 1 | RS | Normal ultrastructure |
| RSPH4A | Radial spoke head protein 4A | RS | Central apparatus defect |
| RSPH9 | Radial spoke head protein 9 | RS | Central apparatus defect |
| HYDIN | Hydrocephalus-inducing protein homolog | CA | Central apparatus defect |

The genetics of PCD has provided unexpected and sometimes surprising insights into ultrastructural phenotypes. For instance, mutations in the dynein axonemal heavy chain 11 (DNAH11) gene (MIM 603339 ) encodes an outer dynein arm protein and, unlike the genes listed previously, is not associated with an ultrastructural defect and cilia have normal (or more rapid) beat frequency. It is likely that an abnormal waveform leads to inefficient mucociliary clearance. Mutations in CCDC39 (MIM 613798 ) and CCDC40 (MIM 613799 ), genes that are essential to assembly of the nexin-dynein regulatory complex and inner dynein arms, yield inconsistent ultrastructural abnormalities characterized by absent inner dynein arms in all axonemes and misplaced radial spokes, and microtubular disorganization in only 10%–20% of cilia. Investigations have shown that CCDC39 and CCDC40 serve as “rulers” and are critical for the construction of the ciliary scaffold and accurate spacing of the radial spokes along the axoneme.
More recently, several patients who have mutations in genes encoding cyclin O (CCNO) and multiciliate differentiation and DNA synthesis–associated cell cycle protein (MCIDAS) were found to have symptoms consistent with PCD and markedly reduced motor cilia on the epithelial surface. CCNO is required for centriole production, and mutations cause mislocalization of basal bodies, consistent with defective basal body replication and migration to the cell surface. MCIDAS controls centriole replication required for normal ciliated cell formation. Thus, although the absence of epithelial cilia frequently can be sequelae of infectious insults, it may also have a genetic etiology.
Although the genetic testing holds considerable promise as a diagnostic tool for PCD, it has limitations. First, because PCD is an autosomal recessive condition, presence of a single mutation (or single mutations in different genes) is insufficient to make the diagnosis. Whole exome or genome sequencing is a powerful tool that has allowed more rapid identification of sequence variants, but it is not always clear whether these variants are disease causing. Polymorphisms are fairly common, especially in larger PCD-associated genes, like the heavy chain dyneins. Irrespective of the gene affected, nonsense mutations or deletions result in a truncated protein and loss of function and are likely pathogenic. However, rare sequence variants and missense mutations that change a single amino acid are more difficult to link to disease. In addition, it is possible that some biallelic variants may not cause classic PCD but result in mildly reduced cilia function and more subtle clinical manifestations.
Ultrastructural Ciliary Defects
The ciliated epithelium of patients with PCD may appear normal, but distinct ultrastructural defects of the ciliary axoneme have been identified of cilia using transmission electron microscopy, including abnormalities in the dynein arms, radial spokes, nexin links, and microtubular organization ( Fig. 71.6 , Table 71.2 ). These distinct ultrastructural defects are generally apparent in cilia throughout the airways, middle ear, and oviduct, as well as in sperm flagella, demonstrating that PCD is a generalized disorder of genetic origin.

| DYNEIN ARM DEFECTS |
|
| RADIAL SPOKE DEFECTS |
|
| MICROTUBULAR TRANSPOSITION DEFECTS |
| Absent central pair of tubules with transposition of outer doublet(s) |
| OTHER |
|
Dynein abnormalities occur in approximately 80% of PCD patients who have ultrastructural defects and include complete absence of both inner and outer dynein arms, complete or partial absence of outer dynein arms alone, and in rare cases absence of inner dynein arms alone. Outer dynein arms are readily identifiable, but inner dynein arm analyses can be difficult because these structures are often obscured by abundant overlying electron-dense material. Computer-assisted image analysis has been used to improve visualization of inner dynein arms, thereby enhancing the ability to detect inner arm defects. Because inner dynein arms abnormalities may be observed in normal subjects with recent respiratory infections or other epithelial insults, the diagnosis of PCD in individuals with only inner dynein arm defects can be challenging and repeat confirmatory testing is recommended.
A number of defects in radial spokes have been associated with ciliary dysmotility, including total absence of radial spokes and absence of radial spoke heads. These defects are recognized by an eccentric position of the central pair of microtubules that are normally stabilized in a central position by the radial spokes. Central displacement of one of the outer microtubular doublets may also occur. A characteristic alteration in the 9+2 configuration of axonemal microtubules has been described in some families with PCD. Typically, the central pair of tubules is missing, and one of the outer microtubular doublets is transposed to the center in both cilia and flagellae.
Acquired ultrastructural defects have been described that are likely secondary to epithelial insults, such as viral infections. Indeed, ciliary disorientation was initially described as an ultrastructural phenotype of PCD, but this defect is more likely related to airway injury than a genetic defect. One approach to discern whether defects are acquired or congenital is to examine ciliary ultrastructure in epithelial cell cultures grown at air-liquid interface, which allows cells to polarize and redifferentiate.
It is important to note that approximately 30% of patients who have typical clinical manifestations of PCD and ciliary dysmotility will have normal ciliary ultrastructure, indicating that some defects affect function but not structure. As mentioned previously, some ciliary abnormalities in PCD are not found in every axoneme and the changes are often attributed to an acquired defect, further complicating the use ciliary ultrastructural analyses as a diagnostic tool.
Functional Ciliary Defects
In contrast to cystic fibrosis, in which abnormal apical channels result in obstruction and persistent infection, the ineffective cilia beat leads to impaired mucociliary clearance and chronic infections in the PCD airway ( Fig. 71.7 ). Advances in high-speed, high-resolution videomicroscopy have led to its development as a diagnostic tool for PCD, mainly in Europe. Digital imaging of ciliary motion in multiple planes has permitted comprehensive analysis of abnormal beat patterns. Ciliary motion may be assessed by videomicroscopy of freshly excised ciliated epithelium obtained by scrape or brush biopsy of the inferior surface of the nasal turbinate, or by bronchial brush biopsy. Freshly collected ciliated cells maintain ciliary beating for several hours if placed in appropriate culture media. In PCD the cilia may be immotile or dyskinetic. High-speed videomicroscopy has shown that certain beat patterns are associated with specific ultrastructural defects; specifically, absence of outer dynein arms is associated with immotility or a slightly flickering beat pattern; isolated inner dynein arm defects or radial spoke defects are associated with a slow, stiff motion; and ciliary transposition defects are associated with a circular beat pattern that may have a normal beat frequency but lacks the directional bend. Representative high-speed videomicroscopy videos of ciliary motility (provided by Amelia Shoemark at National Heart & Lung Institute, Imperial College, London, recorded at 500 frames per second and playback at 60 frames per second, magnification 100×, temperature 37°C) are available online for healthy control ( ![]() ) and individuals with PCD confirmed by genetic mutations in DNAH5 (
) and individuals with PCD confirmed by genetic mutations in DNAH5 ( ![]() ), and DNAH11 (
), and DNAH11 ( ![]() ).
).




