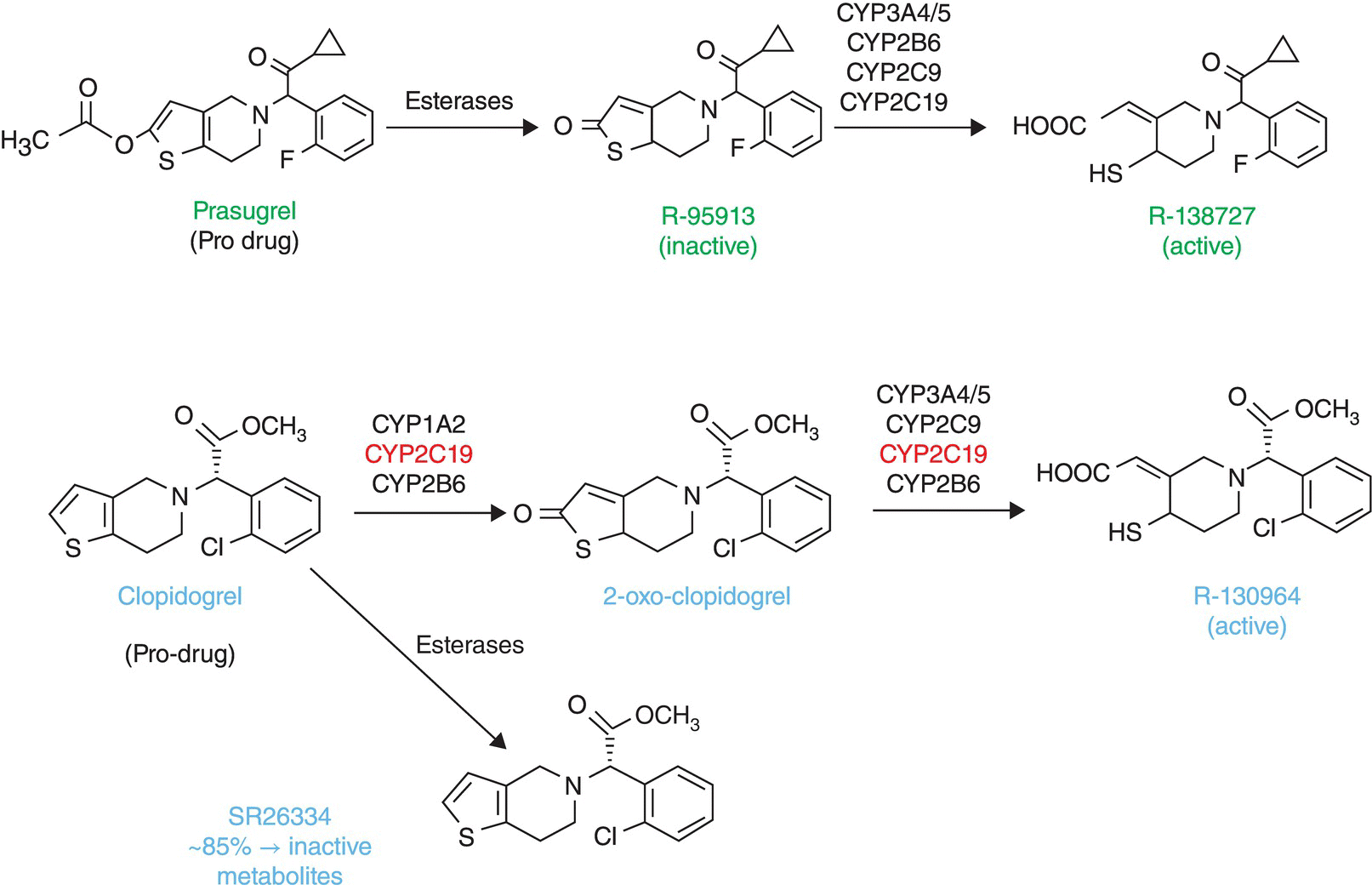
20 Christoph Varenhorst1,2, Anna Oskarsson1, and Stefan James1,2 1 Uppsala Clinical Research Center, Uppsala, Sweden 2 Uppsala University Hospital, Uppsala, Sweden The thienopyridines (e.g., clopidogrel, prasugrel) are metabolized by cytochrome P450 (CYP450) in the liver to active metabolites that irreversibly bind to the P2Y12 receptor, causing inhibition of platelet activation. Clopidogrel efficacy is hampered by a number of factors and poor responsiveness to clopidogrel occurs in 20–30 % of patients [1]. This has prompted the development of antiplatelet drugs, such as prasugrel, that inhibit platelets more effectively. Clopidogrel’s and prasugrel’s conversions to the active metabolite have some key differences. Clopidogrel is metabolized by two CYP-dependent pathways. Prasugrel, on the other hand, first undergoes a rapid de-esterification in the intestine to an intermediate metabolite, which then is converted to the active metabolite in one CYP-dependent step [2] (Figure 20.1). The simpler activation for prasugrel results in a more potent, faster acting, and less variable platelet inhibition compared to clopidogrel. Figure 20.1 Prasugrel and clopidogrel metabolism. (Source: Adapted from Farid, N.A., Payne, C.D., Ernest, C.S. 2nd. et al. (2008) Prasugrel, a new thienopyridine antiplatelet drug, weakly inhibits cytochrome P450 2B6 in humans. Journal of Clinical Pharmacology, 48 (1), 53–59. Reproduced with permission of John Wiley & Sons Ltd.) Interaction studies indicate that the conversion to the prasugrel active metabolite is not influenced by reduced function polymorphisms, whereas the active metabolite formation for clopidogrel is at least affected by CYP2C19 polymorphisms [3, 4]. Also, prasugrel does not seem to interact to any clinical relevant extent with other drugs, including those CYP450 isoenzymes involved in prasugrel metabolism (CYP3A4, CYP2C9, CYP2C19, and CYP2B6) [2, 5]. Both the loading and maintenance doses of prasugrel result in a more rapid, consistent, and greater platelet inhibition than clopidogrel [1]. After a loading dose (LD) of 60 mg prasugrel, a maximum 60–70 % platelet inhibition is reached within 2–4 h in patients with stable coronary artery disease. Maintenance dosing with 10 mg prasugrel once daily achieves an average of 50 % platelet inhibition [6, 7]. However, in ST-segment elevation myocardial infarction (MI) patients undergoing primary PCI, the drug onset seems to be slower, with up to 4 h to achieve adequate platelet inhibition [8]. A possible explanation for the difference in time to drug-effect onset is altered gastrointestinal absorption in the acute phase of coronary syndromes. After the prasugrel treatment is discontinued, the platelet inhibition decreases to pretreatment levels within 7–10 days [9]. The SWAP (Switching Anti Platelet) study randomized patients who had received a run-in period of clopidogrel 75 mg once daily to a regimen of placebo LD/clopidogrel 75 mg maintenance dose (MD) or placebo LD/prasugrel 10 mg MD or prasugrel 60 mg LD/prasugrel 10 mg MD. Platelet reactivity was significantly lower with prasugrel maintenance dosing compared to clopidogrel maintenance dosing (41.1 % vs. 55.0 % inhibition of platelet aggregation (IPA); p < 0.0001) and prasugrel loading/maintenance dosing compared to clopidogrel maintenance dosing (41.0 % vs. 55.0 % IPA; p < 0.0001). The SWAP study confirmed that switching from clopidogrel to prasugrel leads to a further reduction in platelet reactivity by 1 week using prasugrel maintenance dosing or within 2 h with the administration of a prasugrel LD [10]. The pivotal TRITON-TIMI (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel – TIMI) 38 was the first mega trial assessing the hypothesis that higher and less variable P2Y12 platelet inhibition would prevent clinical ischemic events [11]. The trial was preceded by two phase II studies assessing the pharmacodynamics and safety of prasugrel. Although there was no significant difference in TIMI major and minor non-CABG-related bleeding in these studies, access site bleeding was more frequent with prasugrel compared to clopidogrel [12]. The TRITON-TIMI 38 enrolled 13,608 patients with moderate- and high-risk ACS and planned to PCI. Patients were randomized to prasugrel (60 mg LD and 10 mg daily MD) or clopidogrel (300 mg LD and 75 mg daily MD) for a median time of 14.5 months [11]. Prasugrel was associated with a significant reduction of the main efficacy end point (cardiovascular death, nonfatal MI, or nonfatal stroke), with an event rate of 12.1% in the clopidogrel group versus 9.9% in the prasugrel group (p < 0.001). This was mainly driven by a reduction in MI and stent thrombosis; no difference was observed in mortality. Study drugs were not given until the coronary anatomy was known. It has therefore been suggested that this design favored prasugrel over clopidogrel in preventing stent thrombosis by countervailing optimal upstream dosing. The reduction of ischemic end points with prasugrel was accompanied by a higher incidence of major bleeding (TIMI major bleeding not related to CABG), occurring in 1.8% of the patients in the clopidogrel group versus 2.4% in the prasugrel group (HR 1.32; 95% CI 1.03–1.68; P < 0.05). Post hoc
Prasugrel
Introduction
Mechanism of action
Prasugrel pharmacodynamics
Clinical studies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


