8 Platelet Inhibitor Agents
 Basic Principles of Anti-Platelet Therapy
Basic Principles of Anti-Platelet Therapy
Platelets have a key role in normal hemostasis and in the pathogenesis of atherothrombotic disease. Platelets provide an initial hemostatic plug at the site of vascular injury and promote pathophysiologic thrombosis, which, in turn, precipitates myocardial infarction (MI), stroke, and peripheral vascular occlusions. Therefore, anti-platelet agents are key in cardiovascular disease management. In particular, the goal of anti-platelet treatment strategies is to reduce the risk of recurrent atherothrombotic events without excessive bleeding complications. However, because both pathologic and physiologic functions of platelets are caused by the same mechanism, it is difficult to separate therapeutic benefits from potential harmful effects. Platelet plug formation at sites of vascular injury occurs in three stages: (1) initiation phase, which involves platelet adhesion; (2) extension phase, which includes activation, additional recruitment, and aggregation; (3) perpetuation phase, characterized by platelet stimulation and stabilization of clot.1 Circulating platelets are quiescent under normal circumstances and do not bind to the intact endothelium. However, endothelial damage leads to the exposure of circulating platelets to the subendothelial extracellular matrix and triggers platelet recruitment and adhesion (Fig. 8-1).2 In the initial phase of primary hemostasis, the tethering of platelets at sites of vascular injury is mediated by GP Ib-IX-V receptor complex, which binds von Willebrand factor (vWF). Subendothelial collagen exposed by damaged vessel engages platelets via GP VI and GP Ia/IIa receptors. These interactions allow the arrest and activation of adherent platelets. In the extension phase, additional platelets are recruited and activated via soluble agonists. These platelet-activating factors include adenosine diphosphate (ADP), thromboxane A2 (TXA2), epinephrine, serotonin, collagen, and thrombin. Signaling via ADP receptors contributes to platelet activation during both protective hemostasis and pathologic thrombosis. Two ADP receptors are expressed by platelets: P2Y1 which couples to Gαq and contributes to initial aggregation, and P2Y12 which couples to Gα12 and decreases cyclic adenosine monophosphate (cAMP), stabilizing the platelet aggregate.3 P2Y12 receptor signaling also stimulates surface expression of P-selectin and secretion of TXA2. TXA2 is produced de novo and, like ADP, is released from adherent platelets. It is generated from arachidonic acid through conversion by cyclo-oxygenase 1 (COX-1) and thromboxane synthase. TXA2 binds platelet receptors TPα and TPβ; however, its effects in platelets are mediated primarily through TPα. ADP and TXA2 are secreted from adherent platelets and contribute to the recruitment of circulating platelets and promote alterations in platelet shape and granule secretion. Thus platelet activation is amplified and sustained during the extension phase. Thrombin, generated at the site of vascular injury, represents the most potent platelet activator.4 Thrombin contributes to the formation of the hemostatic plug and platelet thrombus growth. Thrombin also directly activates platelets through stimulation of the protease-activated receptors (PARs). Human platelets express two PARs for thrombin: PAR1 and PAR4. Thrombin facilitates the production of fibrin from fibrinogen, contributing to the formation and stabilization of the hemostatic plug.4 The final common pathway is activation of the integrin GP IIb/IIIa, which allows platelets to bind fibrinogen with high affinity, leading to platelet aggregates.5 In the perpetuation phase, the platelet-rich thrombus and coagulation cascades reinforce one another and culminate in the generation of a stable platelet-fibrin–rich plug at the sites of injury.
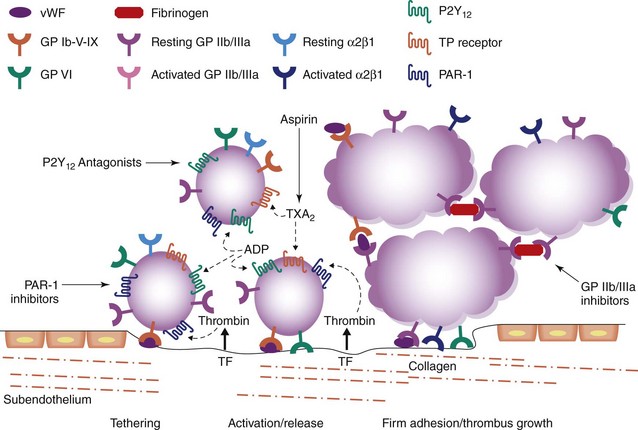
Figure 8-1 Platelet mediated thrombosis.
(Adapted from Varga-Szabo D, Pleines I, Nieswandt B: Cell adhesion mechanisms in platelets, Arterioscler Thromb Vasc Biol 28:403–412, 2008.)
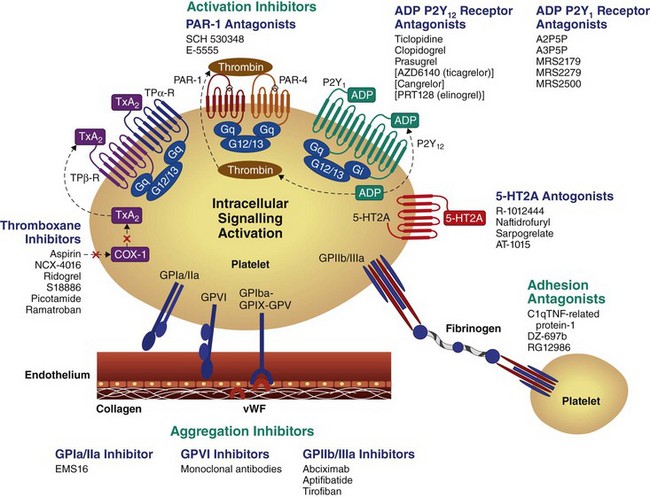
The mechanisms by which anti-platelet drugs interfere with platelet function involve targeting enzymes or receptors that are critical for the synthesis or action of important mediators of these functional responses. Current and investigational oral anti-platelet therapies target key platelet signaling pathways (Fig. 8-2). This chapter reviews the mechanism of action, efficacy and safety of anti-platelet agents inhibiting key platelet-signaling pathways, including the TXA2 pathway, P2Y12 receptor, PAR1 receptor, phosphodiesterase III, and the GP IIb/IIIa receptor, focusing on their roles in PCI.
 Aspirin
Aspirin
Pharmacokinetics and Pharmacodynamics
Aspirin is rapidly absorbed in the stomach and small intestine, achieving peak plasma levels in 30 to 40 minutes. Esterases in the gastrointestinal (GI) mucosa and liver hydrolyze aspirin into salicylic acid, which then interacts with platelets in the portal circulation. The half-life is short, approximately 20 to 30 minutes, but the pharmacodynamic effect is prolonged, given the permanent inactivation of platelet COX-1 activity. Low-dose aspirin requires several days to effectively suppress TXA2 production. A loading dose is needed to quickly achieve effective platelet inhibition in aspirin-naïve subjects. Loading doses greater than 300 mg do not appear to provide additional pharmacodynamic benefit at 2 hours after ingestion. The American College of Cardiology (ACC)/American Heart Association (AHA)/Society for Cardiovascular Angiography and Interventions (SCAI) 2007 PCI guidelines state that patients not already taking daily long-term aspirin therapy should be given 300 mg to 325 mg of aspirin at least 2 and preferably 24 hours before PCI is performed (Class I, Level of Evidence: C).6
Aspirin Dose after Percutaneous Coronary Intervention
Maintenance aspirin regimens of as low as 30 mg daily are adequate to completely inhibit serum TXB2 production (a marker of platelet thromboxane production) in healthy individuals. Collaborative meta-analyses of the clinical benefit of long-term aspirin in high-risk patients show that doses greater than 75 to 150 mg are no more effective in reducing ischemic events but are associated with a greater risk of bleeding. However, patients undergoing PCI and receiving stents are not represented in these studies. The ACC/AHA/SCAI 2007 PCI guidelines state that in patients without allergy or risk of bleeding, aspirin 162 mg to 325 mg daily should be given for at least 1 month after bare metal stent (BMS) implantation, 3 months after sirolimus-eluting stent implantation, and 6 months after paclitaxel-eluting stent implantation, after which daily long-term aspirin use should be continued indefinitely at a dose of 75 mg to 162 mg (Class I, Level of Evidence, B).6 The impact of different aspirin dosages on ischemia and bleeding in stented patients was examined in a post hoc, observational analysis of the PCI cohort of the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial. This analysis suggested that doses of aspirin <200 mg may be optimal after PCI with BMSs.7 The study stratified the 2658 patients who underwent PCI for acute coronary syndrome in the CURE trial into three groups: high-dose (≥200 mg), medium-dose (>100 mg to <200 mg), and low-dose aspirin (≤100 mg).7 There were no differences in the unadjusted or adjusted rates of death, MI, or stroke between groups (high-dose versus low-dose, adjusted HR 1.00 [95% CI 0.67–1.48]; medium-dose versus low-dose, adjusted HR 1.09 [95% CI 0.73–1.60]). Unadjusted and adjusted rates of major bleeding were significantly greater with high-dose aspirin compared with low-dose aspirin (adjusted HR 2.03 [95% CI 1.15–3.57]). The Clopidogrel and Aspirin Optimal Dose Usage to Reduce Recurrent Events—Seventh Organization to Assess Strategies in Ischemic Syndromes (CURRENT–OASIS 7) examined the safety and efficacy of higher-dose aspirin (300 to 325 mg daily) compared with lower-dose aspirin (75 to 100 mg daily) in 25,086 patients with ACS treated with an invasive strategy. All patients received an aspirin loading dose ≥300 mg the day of randomization, and patients were also randomized in a 2-by-2 factorial design to standard-dose or double-dose clopidogrel. In the overall cohort, the rate of cardiovascular death, MI, or stroke at 30 days was not different between higher-dose or lower-dose aspirin (4.2 versus 4.4%, HR 0.97 [95% CI 0.86–1.09], P = 0.61). The incidence of major bleeding as defined by the trial was not different between groups (2.3% vs. 2.3%, HR 0.99 [95% CI 0.84 to 1.17], P = 0.9). Minor bleeding was more frequent with higher-dose aspirin (5.0% versus 4.4%, HR 1.13 [95% CI 100–1.27], P = 0.04), as was GI bleeding (0.4% versus 0.2%, P = 0.04). The findings were similar among those patients who underwent PCI, approximately 42% of whom received a drug-eluting stent, and there was no difference in the incidence of stent thrombosis within the PCI cohort. Therefore, a treatment strategy of lower-dose aspirin in invasively managed patients with ACS for 30 days appears to provide similar ischemic outcomes as higher-dose aspirin with less minor bleeding and less GI bleeding. However, longer treatment durations have not been examined in a randomized fashion.
 P2Y12 Inhibitors
P2Y12 Inhibitors
Thienopyridines
The thienopyridines—ticlopidine, clopidogrel, and prasugrel—are pro-drugs that require biotransformation into an active metabolite to exert their anti-platelet effect. The active metabolite irreversibly binds and antagonizes the P2Y12 receptor for the platelet’s lifespan (7 to 10 days). Differences in the rapidity and magnitude of platelet inhibition between the thienopyridines are predominantly the result of differences in pro-drug metabolism that affect the efficiency of active metabolite formation. Since the interaction between the active metabolite and the P2Y12 receptor is irreversible, a substantial waiting period for platelet functional recovery is required after thienopyridine exposure, which appears to be related to the magnitude of the initial inhibition.8,9
Ticlopidine
Ticlopidine was the first thienopyridine to be introduced into clinical practice. It has a slow onset of action, is poorly tolerated, and its use is associated with blood dyscrasias. The incidence of neutropenia has been reported to be 2.4%, peaking at 4 to 6 weeks after the start of therapy; the incidence of aplastic anemia is 1 in 4000 to 8000 patients; and the incidence of thrombotic thrombocytopenic purpura is approximately 1 in 2000 to 4000 patients. The onset of hematologic disorders is rare after 3 months of therapy. Therefore, hematologic monitoring is required before initiation and for the first 3 months of exposure. The Stent Anticoagulation Re-stenosis Study (STARS) demonstrated that the combination of aspirin and ticlopidine significantly reduced the rate of death, angiographically evident stent thrombosis, MI, or revascularization at 30 days by 85% compared with aspirin alone and by 80% compared with the combination of aspirin and warfarin.10 Ticlopidine has been widely replaced by clopidogrel, given its better tolerability and lack of blood monitoring requirements.
Clopidogrel
Metabolism
Clopidogrel is a pro-drug that requires hepatic conversion into an active metabolite to exert its anti-platelet effect (Fig. 8-3). Approximately 85% of absorbed clopidogrel is hydrolyzed by human carboxylesterase1 in the liver into an inactive carboxylic acid metabolite, so only a fraction of absorbed clopidogrel is available for conversion into the active metabolite by the cytochrome P450 (CYP) system. Hepatic biotransformation of absorbed clopidogrel into the active metabolite is thought to occur through a two-step process. The thiophene ring of clopidogrel is first oxidized to 2-oxo-clopidogrel, which is then hydrolyzed to a highly labile active metabolite (R-130964) that forms a disulfide bond with the P2Y12 receptor as platelets pass through the liver. The first metabolic step involves the isoenzymes CYP2C19, CYP2B6, and CYP1A2, and the second step involves the isoenzymes CYP2C19, CYP2B6, CYP3A4, CYP3A5, and CYP2C9. An alternative pathway for the oxidative biotransformation of clopidogrel that does not involve CYP2C19 has been suggested.11 In this formulation, CYP-catalyzed oxidation of clopidogrel to 2-oxo-clopidogrel is mediated by CYP3A, CYP2B6, and CYP1A2, and conversion of 2-oxo-clopidogrel to the thiol active metabolite is mediated by the esterase, paraoxonase-1 (PON1). The catalytic activity of PON1 is proposed to be the rate-determining step for the active metabolite formation of clopidogrel.
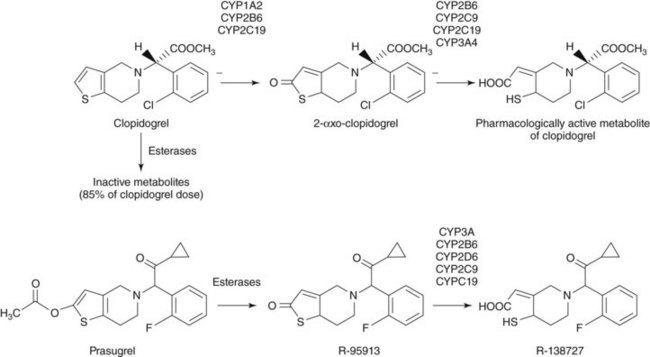
Pharmacodynamics
A daily dose of clopidogrel 75 mg requires 3 to 7 days to reach steady-state platelet inhibition. A loading dose provides a rapid onset of action. However, the clinical benefit of a 300 mg loading dose may not be seen until 6 hours or as long as 15 hours after administration.12,13 Larger doses provide higher circulating levels of active metabolite, more rapid onset, and more intense inhibition.14,15 Peak inhibition after a 600-mg loading dose occurs at 4 to 6 hours after exposure.14,16 A 900-mg loading dose may or may not provide more rapid and additional suppression of platelet function compared with 600 mg, as the intestinal absorption of clopidogrel may be limited at doses greater than 600 mg.14,15 A maintenance dose regimen of 150 mg daily is associated with greater inhibition than a dose of 75 mg daily.17,18 There is wide variability among individuals in the anti-platelet effect of clopidogrel after either a loading dose or a maintenance dose. Higher doses of clopidogrel reduce, but do not eliminate, this variability. The pharmacodynamic response to clopidogrel has been associated with CYP2C19 genotype, age, diabetes mellitus, body mass index, gender, ACS presentation, active smoking, renal dysfunction, pretreatment reactivity, and concomitant therapy with calcium channel blockers or proton pump inhibitors (PPIs).19–22 However, clinical characteristics and the CYP2C19 genotype only partly explain the variability in on-treatment reactivity.20,23 The level of ADP-induced platelet reactivity measured by several ex vivo platelet function tests have been associated with clinical outcomes in clopidogrel-treated patients undergoing PCI.24
Clinical Studies
Non–ST Elevation Acute Coronary Syndrome
The longer-term ischemic benefit of clopidogrel in patients presenting with ACS was established by the Clopidogrel in the Unstable Angina to Prevent Recurrent Ischemic Events (CURE) Trial. This trial randomized 12,562 patients presenting with non–ST elevation ACS to aspirin and clopidogrel (300 mg loading dose followed by 75 mg daily) or aspirin alone for 3 to 12 months.25 The composite endpoint of cardiovascular death, nonfatal MI, or stroke occurred in 9.3% of the patients in the clopidogrel group and 11.4% of the patients in the placebo group (P < 0.001). Clopidogrel therapy was associated with an increased rate of major bleeding as defined by the trial (3.7 vs. 2.7%, P = 0.001). In the population of patients enrolled in CURE that underwent PCI (17% of the overall cohort, 82% of whom received a BMS), pretreatment with clopidogrel for a median of 6 days reduced the rate of cardiovascular death, MI, or urgent target-vessel revascularization within 30 days from 6.4% to 4.5% (P = 0.03).
ST Elevation Myocardial Infarction
The Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myocardial Infarction (CLARITY-TIMI)-28 trial randomized 3491 patients ≤75 years of age receiving aspirin and fibrinolytic therapy within 12 hours of an ST elevation myocardial infarction to clopidogrel 300 mg followed by 75 mg daily or placebo. All patients underwent mandated angiography 2 to 8 days later. Clopidogrel significantly reduced the rate of an occluded infarct–related artery, death, or recurrent MI before angiography (15.0% vs. 21.7%, P < 0.001), without increasing TIMI-defined major bleeding, minor bleeding, or intracranial hemorrhage.26 A prespecified analysis of patients who underwent PCI demonstrated that clopidogrel significantly reduced ischemic events from randomization through 30 days, from PCI through 30 days, and from randomization to PCI.27 This trial supports the use of clopidogrel in patients ≤75 years old presenting with ST elevation myocardial infarction (STEMI) and treated with aspirin and fibrinolysis.
Pretreatment for Percutaneous Coronary Intervention
The rationale for clopidogrel pretreatment is based on the slow onset of a substantial pharmacodynamic effect even after a clopidogrel loading dose. The Clopidogrel for the Reduction of Events During Observation (CREDO) trial randomized 2116 patients with stable coronary artery disease (CAD), unstable angina, or recent ACS to a clopidogrel 300 mg loading dose or placebo 3 to 24 hours before PCI. All patients received clopidogrel 75 mg daily for 28 days thereafter; patients in the control arm did not receive a loading dose. Pretreatment did not significantly reduce the primary composite endpoint of death, MI, and urgent target revascularization at 28 days (6.8% vs. 8.3%, P = 0.23). Post hoc analysis suggested that longer durations of pretreatment were associated with improved outcomes, but little benefit was achieved when the treatment duration was less than 12 hours.13 A prospectively planned analysis of the 1863 patients in CLARITY-TIMI 28 undergoing PCI after mandated angiography showed that pretreatment for a median duration of 3 days in patients with STEMI treated with aspirin and fibrinolysis significantly reduced the incidence of cardiovascular death, MI, or stroke following PCI (3.6% vs. 6.2%, P = 0.008) and from randomization through 30 days (7.5% vs. 12.0% P = 0.001).27 Unfortunately, the use of a 300-mg loading dose in CREDO, PCI-CURE, and PCI-CLARITY and the prolonged duration of pretreatment in PCI-CURE and PCI-CLARITY limit their applicability to current practice patterns for both elective and urgent PCIs. The ischemic benefit of a shorter pretreatment duration of high-dose clopidogrel before PCI has not been examined in a large, randomized, placebo-controlled trial. Post hoc analysis of the Intracoronary Stenting and Antithrombotic Regimen-Rapid Early Action for Coronary Treatment (ISAR-REACT) trial, which compared abciximab with placebo in elective PCI patients who were treated with clopidogrel 600 mg for at least 2 hours before intervention, showed no incremental benefit from durations of pretreatment >2 to 3 hours.28 The PRAGUE-8 study randomized 1028 patients undergoing coronary angiography and potential ad hoc PCI for stable angina to either clopidogrel 600 mg >6 hours before angiography or clopidogrel 600 mg in the catheterization laboratory only in the case of PCI.29 There were no differences in the rate of death, MI, stroke, or re-intervention between groups at 7 days, either in the entire population or the subgroup undergoing PCI (0.8% vs. 1.0%, P = 0.7; 1.3% vs. 2.8%, P = 0.4, respectively), but bleeding was increased in the pretreatment group (3.5% vs. 1.4%, P = 0.025). The findings of this small trial support a strategy of “on the table” clopidogrel loading before ad hoc PCI in elective patients, although the findings must be interpreted within the context of the relatively small sample size and very low event rates. The findings are supported by another smaller trial, Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty (ARMYDA) PRELOAD, which randomized 409 patients (39% with acute coronary syndrome) to a 600 mg clopidogrel loading dose 4 to 8 hours before PCI or a 600 mg load given in the catheterization laboratory after coronary angiography and before PCI.30 The rates of major adverse cardiovascular events at 30 days were similar between groups, occurring in 10.8% of patients pretreated compared with 8.8% in the patients receiving clopidogrel in the laboratory (P = 0.7). There were no differences in the rates of bleeding.
Dosing Strategies
Pharmacodynamic studies have demonstrated that higher clopidogrel loading doses and maintenance doses provide more rapid onset of action and greater levels of inhibition compared with a 300-mg loading dose and a 75-mg maintenance dose, respectively.14–1631 Two large randomized studies, CURRENT-OASIS 7 and The Gauging Responsiveness With A VerifyNow Assay–Impact on Thrombosis And Safety (GRAVITAS), have examined the efficacy and safety of higher dose clopidogrel in patients managed invasively or undergoing PCI.
The CURRENT–OASIS 7 trial examined the ischemic benefit of a higher-dose strategy in 25,086 patients with non–ST elevation ACS and ST elevation ACS undergoing an early invasive strategy, of whom 17,263 underwent PCI.32 Before angiography, patients were randomized to receive (1) a 600 mg loading dose, followed by 150 mg daily for 6 days and 75 mg daily thereafter or (2) a 300-mg loading dose followed by 75 mg daily thereafter. Patients were also randomized to high-dose aspirin or low-dose aspirin in a 2-by-2 factorial design. The primary endpoint, a composite of cardiovascular death, MI, or stroke at 30 days, was no different with double-dose clopidogrel or standard-dose clopidogrel (4.2% vs. 4.4%, P = 0.30). A potential interaction was observed with aspirin dosing, where patients receiving double-dose clopidogrel had better outcomes when treated with higher-dose aspirin. This observation must be taken within the context that the interaction P value was 0.04, which did not meet the trial’s prespecified criteria for significance (P = 0.01 to adjust for multiple comparisons) and that a mechanism for this potential interaction is unknown. Major bleeding, as defined by the trial, was significantly greater in the patients randomized to double-dose clopidogrel (2.5% vs. 2.0%, HR, 1.24 [95% CI, 1.05–1.46], P = 0.01), although there were no differences in fatal bleeding, coronary artery bypass graft (CABG)–related bleeding, or TIMI-criteria major bleeding. Within the subgroup of patients who underwent PCI, high-dose clopidogrel was associated with a 13% relative risk reduction in the primary endpoint (3.9% vs. 4.5%, P = 0.04).33 However, the interaction test between patients who did or did not undergo PCI did not reach the prespecified threshold for statistical significance, and therefore the possibility that the results of the PCI subgroup are a chance finding cannot be excluded.32 The GRAVITAS trial tested whether an additional clopidogrel loading dose followed by a 6-month course of clopidogrel 150 mg daily would reduce thrombotic events compared with clopidogrel 75 mg daily in patients who had undergone PCI with a drug-eluting stent (DES) and displayed high on-treatment reactivity according to ex vivo platelet function testing 12 to 24 hours after the intervention. Unlike the population examined by the CURRENT-OASIS 7 trial, the predominant indication for PCI in the enrolled population was stable CAD or low-risk unstable angina. There was no difference in the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months between groups (2.3% vs. 2.3%, P = 0.9). The incidence of severe or moderate bleeding per the Global Utilization of Streptokinase and t-PA for Occluded Coronary Arteries (GUSTO) criteria was not increased with the high-dose regimen (1.4% vs. 2.3%, P = 0.10). The higher-dose clopidogrel regimen had a significant, but only modest, effect on platelet inhibition in patients with high on-treatment reactivity to standard dosing, which may partly explain the similar outcomes of the two groups.
Duration of Therapy
The small but incremental risk of late thrombosis with DESs has raised uncertainty about the optimal duration of dual anti-platelet therapy after PCI. Observational studies have shown that discontinuation of anti-platelet therapy after DES has been associated with late and very late stent thrombosis, but the interpretation of these studies is limited by study design and the presence of potentially unmeasured confounders. Randomized trials from the BMS era demonstrate the benefit of prolonged aspirin and clopidogrel over the first year after PCI.12,34 Patients who have undergone PCI could possibly receive benefits from very long-term clopidogrel because of a reduction in atherosclerosis-mediated events, rather than stent-mediated events. The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial compared aspirin and clopidogrel with aspirin alone over a median treatment duration of 28 months in 15,603 patients with either clinically evident cardiovascular disease or multiple risk factors.35 While there was no difference in the rate of cardiovascular death, MI, or stroke between treatment groups in the overall cohort, a post hoc analysis showed that aspirin and clopidogrel appeared to provide a significant 17% relative risk reduction in the rate of composite ischemic endpoint in patients with a prior MI, ischemic stroke, or symptomatic peripheral arterial disease (PAD).36 In contrast, a randomized study of 2701 patients who underwent DES placement and were free of major adverse cardiovascular or cerebrovascular events at 1 year observed no significant ischemic benefit for extended aspirin and clopidogrel therapy compared with aspirin alone.37 These findings will be confirmed or rebutted by the Dual Antiplatelet Therapy (DAPT) trial (clinicaltrials.gov identifier NCT00977938), which will randomize more than 20,000 patients treated with either a drug-eluting or bare metal stent who are event-free at 12 months post procedure to receive either aspirin and a thienopyridine or aspirin and placebo for an additional 18 months. Current ACC/AHA/SCAI guidelines state that all patients receiving a DES should be given clopidogrel 75 mg daily for at least 12 months if they are not at high risk of bleeding; and for patients receiving a BMS, clopidogrel should be given for a minimum of 1 month, ideally up to 12 months, unless the patient is at increased risk of bleeding, in which case it should be given for a minimum of 2 weeks.6 In the setting of ACS, for post-PCI patients receiving a stent (BMS or DES), a daily maintenance dose should be given for at least 12 months and for up to 15 months unless the risk of bleeding outweighs the anticipated net benefit afforded by a thienopyridine, and continuation beyond 15 months may be considered in patients undergoing DES placement.38
Role of CYP2C19
The anti-platelet effect of clopidogrel is dependent on the generation of an active metabolite through the hepatic CYP450 system. Patients who are carriers for genetic polymorphisms that reduce the catalytic activity of CYP2C19 have lower clopidogrel active metabolite levels and diminished platelet inhibition with treatment. Approximately 5% to 12% of the variability in ADP-induced platelet reactivity appears to be explained by the carriage of the reduced function CYP2C19*2 allele.20,23 The sensitivity of active metabolite generation to changes in the catalytic activity of CYP2C19 may be attributed to the important contribution of this enzyme to both steps in clopidogrel biotransformation. Decreased CYP2C19 function could lead to a bottleneck at the level of hepatic activation, thereby shunting the pro-drug into the pathway leading to an inactive carboxylic acid metabolite.
Predicted Metabolic Phenotype
Patients can be classified on the basis of the predicted metabolic phenotype of the CYP2C19 genotype. The single nucleotide polymorphisms that affect enzyme activity are described using the established “star allele” nomenclature. The CYP2C19*1 allele denotes the lack of known polymorphisms and therefore is considered to be a wild type. CYP2C19*2 is the most common reduced-function allele, with an allelic frequency of approximately 13% in Caucasians, 18% in African Americans, and 30% in Asians. CYP2C19*3 is the second most common reduced-function allele, with an allelic frequency of approximately 10% in Asians but is rare in other ethnicities. Much less common reduced function alleles include *4, *5, *6, *7, *8, and *10. The *17 variant is associated with increased gene transcription and increased catalytic activity of the enzyme. The combination of two alleles (genotype) can be used to predict the metabolic phenotype of a particular individual (Table 8-2). Metabolic phenotype is associated with the pharmacokinetics and pharmacodynamics of clopidogrel. In a study of healthy volunteers, ultra-rapid metabolizers had the highest exposure to active metabolite and the greatest platelet inhibition, and poor metabolizers had the lowest exposure and least platelet inhibition with both loading and maintenance doses.39 The frequency of poor metabolizers is approximately 2% in the Caucasian population.
TABLE 8-2 Classification of Predicted Metabolic Phenotype According to CYP2C19 Genotype
| CYP2C19 Genotype | Predicted Phenotype |
|---|---|
| *17/*17 | Ultra-rapid metabolizer |
| *1/*17 | Ultra-rapid metabolizer |
| *1/*1 | Extensive metabolizer |
| *1/*2–*8 | Intermediate metabolizer |
| *17/*2–*8 | Intermediate metabolizer/unknown |



