PERSISTENT PULMONARY HYPERTENSION OF THE NEWBORN: PATHOPHYSIOLOGY AND MANAGEMENT
Introduction
Persistence of pulmonary hypertension (PPHN) can occur in newborn infants as a complication of birth-related transition from intra- to extra-uterine life. PPHN occurs in approximately 2 per 1000 live births and primarily affects late preterm and term newborn infants. This condition was originally reported in neonates by Gersony et al in 1969 as the persistence of fetal circulation in newborn infants.1 Fox and colleagues subsequently reported cardiac catheterization findings in a group of neonates with perinatal aspiration syndrome and observed supra-systemic pulmonary artery (PA) pressures associated with severe hypoxemia, in the absence of congenital heart disease (CHD).2 They have used the term PPHN in the newborn as a more accurate description of altered hemodynamics in these neonates. PPHN can occur by itself without associated lung disease (primary PPHN) or as a complication of a variety of lung diseases in the neonate. A proper understanding of the normal birth-related transition in pulmonary circulation and factors that lead to failure of this adaptation are necessary for the prevention and treatment of this condition in the neonates.
Successful transition of the newborn to extra-uterine life requires both alveolar ventilation and a rapid decrease in pulmonary vascular resistance (PVR) at birth. Failure of alveolar expansion leads to respiratory distress, while impaired pulmonary vasodilation results in PPHN. The PVR is normally high in the fetus to allow a major portion of the right ventricular output to bypass the non-respiring lungs via the patent ductus arteriosus (PDA) (see Chapter 1). A rapid decrease in PVR at birth is necessary for the nearly 10-fold increase in pulmonary blood flow (PBF), which synchronizes ventilation with perfusion to establish gas exchange function by the lungs.3–5 Several physiological stimuli, including a 6-fold increase in alveolar oxygen tension, clearance of fetal lung fluid, lung distention, and shear stress, interact in a complementary fashion to initiate the drop in PVR. These birth-related stimuli induce the release of vasodilators such as nitric oxide (NO) and prostacyclin (PGI2) from the endothelium (Figure 16.1).5–9 In addition, oxygen also directly activates voltage-gated and Ca2+-activated potassium channels on the vascular smooth muscle to induce relaxation.10–14 However, the mechanisms of oxygen sensing by the PAs and the unique responses of pulmonary vessels to hypoxia and oxygen remain unclear.
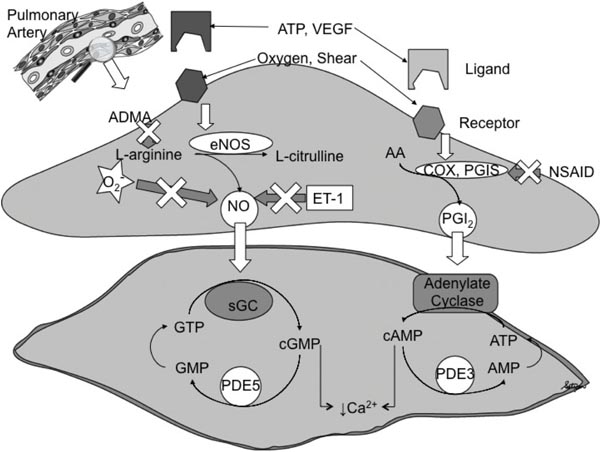
Figure 16.1. Mechanism of endothelium-dependent pulmonary vasodilation at birth. Nitric oxide (NO) and prostacyclin (PGI2) are released in response to birth-related stimuli. NO and PGI2 increase the cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophosphate (cAMP) levels in the smooth muscle cell. Type 5 and type 3 phosphodiesterases (PDEs) degrade these cyclic nucleotides, respectively. A decrease in intracellular Ca2+ levels leads to relaxation of vascular smooth muscle. NO levels are decreased by asymmetric dimethyl arginine (ADMA), superoxide (O2–), and endothelin (ET-1). Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase (COX). AA, arachidonic acid; ATP, adenosine triphosphate; eNOS, endothelial nitric oxide synthase; GMP, guanosine monophosphate; GTP, guanosine triphosphate; PGIS, PGI2 synthase; sGC, soluble guanylate cyclase; VEGF, vascular endothelial growth factor. (Reproduced with permission from Konduri GG, Kim UO. Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn, Pediatr Clin North Am. 2009;56(3):579–600.
Failure to achieve or sustain the normal drop in PVR at birth leads to persistent elevation of PA pressure and severe hypoxemia (PPHN) in the newborn.15–20 Hypoxemia in PPHN results from right-to-left shunting of blood across the patent foramen ovale (PFO) and/or PDA into the systemic circulation. The severity of PPHN in the newborn period encompasses a wide spectrum ranging from a transient elevation in PA pressure to persistent vasoconstriction, and structural remodeling in the PAs that may require prolonged, life-sustaining support in the neonatal intensive care unit (NICU).21 Pulmonary hypertension can contribute significantly to morbidity and mortality in term and preterm babies, and survivors are at an increased risk for long-term neurodevelopmental disabilities.22 The management of infants with moderate to severe PPHN can be complex and requires a prompt and systematic approach. Although the application of inhaled nitric oxide (iNO), a potent pulmonary vasodilator, has been a major advance in the treatment of PPHN, some infants may not respond to iNO and require additional therapies such as extracorporeal membrane oxygenation (ECMO) to survive. Recent advances in basic and translational investigations have increased our understanding of the pathophysiology of PPHN and have led to the development of alternative therapies for managing PPHN. This review outlines our current understanding of the pathophysiology and therapeutic strategies in the management of PPHN.
Physiology of Normal Postnatal Transition
The pulmonary circulation undergoes significant adaptation during fetal and early postnatal life in both structure and vasoreactivity.23–26 The differences in fetal and postnatal circulation are uniquely suited for maximizing oxygen delivery to the systemic organs. In utero, the placenta performs gas-exchange function while the lung is a fluid-filled, non-respiring organ. Therefore, high PVR in the lungs serves the important physiological function of diverting most of the right ventricular output away from the lungs, across the ductus arteriosus (DA) to the descending aorta and the placenta. Fetal lung receives only 8% of the combined cardiac output as a result of this high PVR.23–26 The fetal lung fluid is rapidly reabsorbed at birth and is replaced with air. The PVR in fetal lungs falls quickly at birth, resulting in a 50% decrease in PA pressure in the first few minutes. This dramatic pulmonary vasodilation involves both changes in vascular tone and recruitment of pre-acinar arteries and a gradual remodeling of pulmonary vessels.27 Closure of the PFO and PDA within few hours after birth reduces shunting of blood and improves systemic oxygenation. Several mechanisms including low oxygen tension, low basal production of vasodilators (NO and PGI2), increased levels of vasoconstrictors (endothelin-1 (ET-1) and thromboxane), and proliferation of the vascular smooth muscle cells contribute to the high PVR in utero.28 Owing to the high affinity of fetal hemoglobin (Hb) for oxygen, low oxygen tension (≈20 torr) is well tolerated in utero; however, establishing normal postnatal oxygenation with higher arterial oxygen tension and saturation by the lungs is necessary for the infants to survive.
Regulation of Postnatal Adaptation of Pulmonary Circulation
Postnatal transition is a complex process involving the coordinated regulation of several physiological events that rapidly decrease PVR. These factors are outlined in Table 16.1 and discussed in more detail below.
| Factors maintaining high PVR in the fetus | Factors initiating decrease in PVR at birth |
| Low oxygen tension | Increased oxygen tension |
| Presence of fetal lung fluid | Reabsorption of lung fluid |
| Distension of airspaces | |
| Decreased production of vasodilators—NO and PGI2 | Increased production of vasodilators—NO and PGI2 |
| Increased levels of vasoconstrictors—ET-1, TXA2, and PAF | Decreased production of vasoconstrictors—ET-1, TXA2, and PAF |
| Structural thickening of PAs | Shear stress and thinning of endothelium and vessel wall |
Clearance of Fetal Lung Fluid
The continuous production of fetal lung fluid in utero provides a constant distention of the lung, which is critical for lung parenchymal and vascular development. The fetal lung fluid is secreted by an active process in the epithelial lining of the developing lung via Na+–K+–2Cl− co-transport, which secretes Cl− and is driven by Na+–K+ adenosine triphosphatase (ATPase).29–31 Studies in mice demonstrated that lung distention increases the expression of vascular endothelial growth factor (VEGF) and angiopoietin-1, critical signaling proteins for vascular development.32 The increase in epinephrine and cortisol production during late gestation induces epithelial sodium channel (ENaC) and facilitate fetal lung fluid clearance. Loss of fetal lung fluid early in gestation, as seen in oligohydramnios, predisposes to hypoplasia of the lung, decreased growth of pulmonary vessels, and pulmonary hypertension.33,34 In contrast, clearance of fetal lung fluid acutely at birth contributes to increased PBF during this transition. Studies in near-term fetal sheep have shown that a reduction in fetal lung fluid volume similar to that at birth by itself causes a 3- to 4-fold increase in blood flow to the lungs.30
Postnatal Oxygen Exposure
Oxygen is the most important stimulus for perinatal pulmonary vasodilation. In utero, low oxygen tension (≈20 torr) is one of the key factors that maintain high PVR. Exposure of fetal lung to oxygen at birth (alveolar PO2 ≈ 150 mmHg in room air) induces pulmonary vasodilation.35–37 Studies in fetal lambs have shown that a 2-fold increase in fetal PaO2 alone, in the absence of ventilation, can reproduce the pulmonary vasodilation seen at birth.36 Increased oxygen tension stimulates the activity of both endothelial nitric oxide synthetase (eNOS) and cyclooxygenase (COX) to increase the release of NO and PGI2 from the endothelium (Figure 16.1).38–40 Response of pulmonary circulation to O2 is developmentally regulated; with advancing gestation, pulmonary circulation in the fetus becomes progressively more responsive to the vaso-constrictive effects of hypoxia and vasodilator effect of O2.35,36,41 Doppler studies in pregnant mothers demonstrated that maternal hyperoxia increases PBF in fetuses > 31 weeks of gestation.42
Shear Stress
Increased blood flow into the lungs following reduction in PVR also distends the vasculature and generates shear stress. The PA endothelial lining is particularly sensitive to hemodynamic shear stresses acting on the luminal surface in the direction of blood flow. Shear stress promotes adaptive vasodilatation and structural reorganization of the pulmonary vessels via flattening of the endothelium and thinning of smooth muscle cells and the matrix. Shear stress also induces eNOS expression and activity, which further increases the blood flow.43
Nitric Oxide
Several studies have shown that NO is a critical mediator of pulmonary vasodilation during postnatal transition (Figure 16.1). Oxygen stimulates NO release in part by increasing oxidative phosphorylation and adenosine triphosphate (ATP) release by fetal red blood cells, which induces eNOS activity in the endothelium.44,45 NO also plays a critical role in the pulmonary vascular development in response to VEGF.46,47 NO is both upstream and downstream of VEGF and provides the crosstalk to facilitate the coordinated growth of alveolar and vascular compartments in the fetal lung.47 Chronic inhibition of VEGF has been shown to down-regulate eNOS expression.47 Endothelial NOS protein is developmentally regulated; eNOS protein is present early in gestation and increases with advancing gestational age in utero.48 Increase in eNOS expression and function at term are critical to the rapid transition at birth since unlike the surfactant, NO is not stored in the lung. Therefore, increased expression of eNOS protein is needed to rapidly increase the synthesis and release NO at birth.46 The effects of NO are primarily mediated via activation of soluble guanylyl cyclase (sGC) and increase in cyclic guanosine monophosphate (cGMP) levels in the vascular smooth muscle cell (Figure16.1).49–53
Soluble GC converts guanosine triphosphate (GTP) to cGMP, which activates protein kinase G. This leads to a decrease in Ca2+ influx and vascular smooth muscle relaxation (Figure 16.1).51,52 The vasodilator effect of NO is regulated by a specific phosphodiesterase (PDE) 5 that hydrolyze cGMP. In the fetal lung, PDE5 concentration is high, contributing to the maintenance of high PVR in fetal life52,53 and rapidly falls after birth. Increased expression and activity of PDE5 were reported in animal models of PPHN and contribute to impaired vasodilation at birth.54,55 Endothelial NOS activity is also normally facilitated through its interaction with a constitutively expressed chaperone molecule, HSP90 and a required cofactor, tetrahydro-biopterin (BH4). Konduri et al demonstrated that impaired eNOS–HSP90 association reduces eNOS activity and NO production and contributes to impaired relaxation of PAs in PPHN lambs.56 Depletion of BH4 also contributes to impaired eNOS function and angiogenesis in PPHN.57 Inhibition of eNOS increases pulmonary arterial pressure and attenuates pulmonary vasodilation at birth.57 In addition, expression and activity of eNOS are decreased in fetal lamb model of PPHN and in neonates with PPHN.58–60 Pulmonary vascular tone is also regulated by other NOS isoforms besides eNOS. Selective inhibition of neuronal NOS in animals increases basal PVR and attenuates NO-mediated pulmonary vasodilation at birth.
Prostacyclin (PGI2)
The endothelium also synthesizes prostaglandins (PGs) through the activity of COX-1 and -2, the rate-limiting steps in PG synthesis (Figure 16.1).61 PGI2 synthase converts the intermediate PG, PGH2 to PGI2, the most potent of the vasodilator PGs. PGI2 activates adenylate cyclase, which converts ATP to cyclic adenosine monophosphate (cAMP) in the smooth muscle. Increased intracellular cAMP levels decrease Ca2+ influx and result in vascular smooth muscle relaxation (Figure 16.1). Type 3 phosphodiesterase [PDE-3] degrades cAMP and limits the duration of vasodilation in smooth muscle cells (Figure 16.1). PGI2 synthesis is developmentally regulated; in late gestation, expression of COX, the rate-limiting step in PG synthesis, is increased in the PAs of fetal lambs.61 The vasodilatory effects of PGI2 complement those of NO and are activated by oxygen and ATP, same physiological stimuli that increase NO release (Figure 16.1).
Endothelium-independent Vasodilator Signals
Atrial natriuretic peptide (ANP) is released primarily from atrial myocytes in response to the stretch on the left atrium from increased pulmonary venous return during this transition. ANP increases cGMP levels in vascular smooth muscle cells through the activation of particulate guanylate cyclase (pGC, also known as natriuretic peptide receptor A, NPRA); this system complements eNOS–cGMP pathway and has been shown to also regulate vascular oxidative stress through cGMP generation independent of NO.62 Oxygen has also been shown to directly activate the voltage-gated (Kv) and Ca2+ activated potassium channels (KCa2+ or Maxi K channels) in the vascular smooth muscle.13,63 Activation of these K+ channels leads to hyperpolarization of cell membrane and smooth muscle cell relaxation. A decrease in the expression and activity of Kv channels was observed in animal models of PPHN, although its significance in neonates with PPHN remains unclear.64
Pathophysiologic Mechanisms of PPHN
Increased PVR in babies with PPHN can result from several neonatal disorders that induce vasoconstriction and medial hypertrophy of the PAs. In PPHN, the PAs exhibit endothelial dysfunction, leading to decreased production of vasodilators (NO, PGI2) and increased synthesis of vasoconstrictors such as thromboxane A2 (TXA2) and ET-1. Evidence for these alterations in PPHN is discussed below.
NO-cGMP Signaling
Altered eNOS expression and activity as well as NO release by the vascular endothelium contribute to impaired vasodilation in PPHN. Decreased NO metabolites in the urine and decreased gene expression of eNOS in the umbilical vein endothelial cells were reported in infants with PPHN.60 Studies in animal models have shown that excess production of reactive oxygen species (ROS), such as superoxide and hydrogen peroxide in vascular cells also contribute to the altered production of and unresponsiveness to NO in PPHN.55–57 Oxidative stress can decrease the availability of cofactors for NO synthesis, particularly BH4. Decreased levels of BH4 and accumulation of its oxidation product, dihydro-biopterin (BH2), lead to eNOS uncoupling and generation of superoxide instead of NO from eNOS.57 The downstream mediators of NO signaling are also impaired in PPHN. PDE5 expression and enzyme activity are increased, leading to increased degradation of cGMP and impaired relaxation of smooth muscle cells in PPHN.55,65 ADMA, an endogenous arginine analog, also affects the availability and interaction of arginine with NOS. ADMA inhibits NO release and also causes eNOS uncoupling. ADMA levels are increased in animals and patients with pulmonary hypertension.66,67
Endothelin-1
ET-1 is a potent pulmonary vasoconstrictor secreted by endothelial cells. Infants with PPHN have been reported to have higher ET-1 levels in their blood compared with healthy newborns.68 ET-1 is an important regulator of pulmonary vascular smooth muscle tone during fetal life and acts via ET-A and B receptors.69 ET-A is localized mainly to the endothelium and produces vasodilation, whereas ET-B is localized to the vascular smooth muscle and produces vasoconstriction on interaction with ET-1.69 Since ET-1 acts on 2 receptors with opposing effects, it produces a biphasic effect, with an initial transient vasodilation and a delayed but progressive increase in PVR. In disease states, ET-1 produces mainly vasoconstriction via interaction with ET-B receptor.70–72 A number of studies have shown that NO and ET-1 regulate each other in a reciprocal manner. NO decreases ET-1 production, whereas, ET-1 increases superoxide production and decreases NO bioavailability.71 ET-1 receptor antagonists have been studied primarily in adult pulmonary hypertension with some case reports and pilot studies of their use in neonatal PPHN.73,74
Oxidative Stress
Oxidative stress results when there is an imbalance between ROS production and antioxidant levels. ROS can cause vasoconstriction directly and indirectly by reacting with NO to form peroxynitrite, a potent signaling agent that increases vascular tone. Sources of ROS in vascular cells include increased nicotinamide adenine dinucleotide phosphate, reduced (NADPH) oxidase activity, eNOS uncoupling, xanthine oxidase, and mitochondrial respiration.75–77 Increased ROS production from NADPH oxidase, uncoupled eNOS, and the mitochondria has been demonstrated in PPHN models.78,79 Antioxidant defenses, particularly superoxide dismutase (SOD) system plays a major role in detoxifying the cells of superoxide. Three SOD isoforms are known to be present in vascular cells, cytosolic Cu-Zn SOD (SOD1), mitochondrial localized manganese SOD (SOD2), and extracellular SOD (SOD3). In the fetal lamb model of PPHN, SOD2 expression and activity are downregulated and contribute to impaired pulmonary vasodilation in PPHN.79 This suggests that SOD2 may play an important role in the regulation of pulmonary vasodilation. Since NO is rapidly quenched by superoxide, NO bioavailability partly depends on cellular antioxidant defenses. Scavenging ROS by instillation of intratracheal Cu/Zn-SOD has been shown to improve postnatal pulmonary vasodilation and response to iNO in newborn lambs with PPHN.80 Decreasing oxidative stress prenatally using antenatal betamethasone also improves the postnatal pulmonary vasodilation and antioxidant levels in newborn lambs with PPHN.81 These studies demonstrate the mechanistic role of oxidative stress in the development of increased PVR in PPHN.
Antenatal Drug Exposure
Evidence suggests that perinatal exposure to non-steroidal anti-inflammatory drugs (NSAIDs) and selective serotonin reuptake inhibitors (SSRIs) may be associated with PPHN.82–88 Biological susceptibility to the adverse effects of these agents remains undefined. However, it is an important factor, although not all babies exposed to NSAIDs or SSRIs develop PPHN. The mechanistic link between NSAIDs and PPHN relates to the contribution of PGs in maintaining the patency of DA in fetal life.85 NSAIDs inhibit PG synthesis by inhibiting COX, a rate-limiting enzyme in PG synthesis, which causes premature closure of the DA. Recently, an epidemiologic study suggested an increased incidence of PPHN with prenatal exposure to SSRIs during the third trimester of pregnancy.86,87 The mechanism of the effect of SSRIs on the fetal pulmonary circulation requires further investigation. Preliminary data in a fetal lamb model suggest that serotonin and SSRIs are vasoconstrictors and the serotonin pathway is up-regulated in PPHN.88
Growth Factors and Vascular Remodeling
Thickening of all 3 layers of pulmonary vessels is a consistent histologic finding in the experimental models and infants with severe PPHN at postmortem.89 Increased smooth muscle cell proliferation and extension into non-muscular intra-acinar arteries contribute to increased reactivity of pulmonary vessels to hypoxia, acidosis, and other environmental factors such as hypothermia and ambient noise. The intima may show increased fibroblast infiltration while the adventicia may show increased collagen and elastin deposition. Mediators including ET-1, platelet-activating factor, ROS, platelet-derived growth factor, transforming growth factor (TGF), and other growth factors promote remodeling of pulmonary vessels. NO and PGI2 normally inhibit excessive smooth cell proliferation and vascular remodeling, and their deficiency contributes to this remodeling.90
Prevalence
PPHN affects 1 in every 500 live births in term and near-term (>34 weeks) infants, which equals an estimated 7400 cases per year in the United States.16,21 While PPHN is frequently seen in term and late preterm infants, premature infants <34 weeks of gestation may also occasionally show clinical and echocardiographic features of PPHN.
Classification and Causes
PPHN may be primary or ‘idiopathic’ with no known associated lung disease or secondary to parenchymal lung diseases including respiratory distress syndrome (RDS), meconium aspiration syndrome (MAS), pulmonary hypoplasia resulting from congenital diaphragmatic hernia (CDH) and oligohydramnios, pneumonia, sepsis, and perinatal asphyxia (Figure 16.2). Rare but lethal causes of PPHN are alveolar-capillary dysplasia (ACD) and inborn defects in surfactant pathway, including surfactant protein B (SP-B) and ATP-binding cassette protein member A3 (ABCA3) deficiency.91–93 Important causes of primary/idiopathic pulmonary hypertension are chronic intra-uterine hypoxia, fetal exposure to drugs that causes in-utero ductal closure, such as NSAIDs or SSRIs (which are used as antidepressants).82,86 Lung diseases associated with PPHN include MAS, RDS, pneumonia/sepsis, and lung hypoplasia (Figure 16.2). The distribution of lung diseases associated with PPHN has evolved over time. MAS has been the most common lung disease associated with PPHN, followed by primary PPHN, RDS, and pneumonia or sepsis (Figure 16.2). The incidence of MAS has declined over the last 15 years at most centers with the decrease in post-term pregnancies. Over the same period, incidence of PPHN due to RDS has increased with increased number of late preterm births. Yoder et al reported a 4-fold decrease in the incidence of MAS along with decrease in deliveries past 41 weeks from early to late 1990s.94,95 Over the same period, they reported an increase in the incidence of late preterm births.95
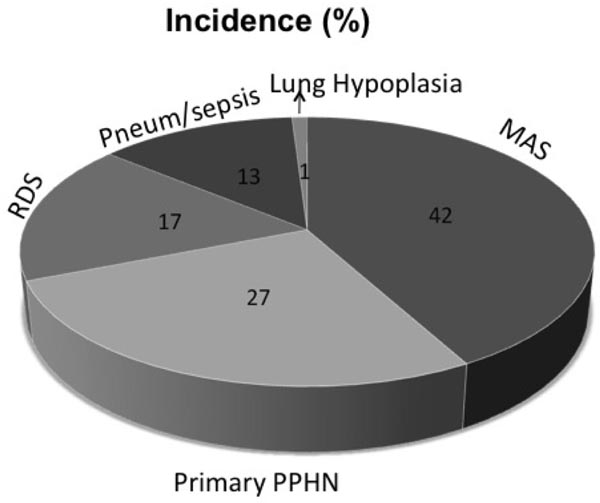
Figure 16.2. Lung diseases associated with PPHN in 299 neonates enrolled in the early iNO study in term and near-term neonates. MAS was the most frequent diagnosis, followed by primary PPHN, RDS, and pneumonia (Pneum) or sepsis. Infants who had congenital diaphragmatic hernia were excluded from this study. Numbers inside each pie represent the percentage of neonates with that diagnosis. MAS, meconium aspiration syndrome; PPHN, persistence of pulmonary hypertension; RDS, respiratory distress syndrome. (Reproduced with permission from Konduri GG, Kim UO. Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn. Pediatr Clin North Am. 2009;56(3):579–600.)
Lung Diseases Associated with PPHN
The pathophysiology, management, and outcome in PPHN are partly related to the lung diseases associated with PPHN. MAS may lead to impairment of alveolar ventilation due to inactivation of surfactant by meconium. Meconium also causes airway obstruction and incites an inflammatory response with release of cytokines. Levels of ET-1 and thromboxane are increased in the lungs in MAS and may contribute to PPHN. The overall decline in the severity and incidence of MAS in the recent times indicates that in-utero stress in post-term pregnancies probably contributed to pulmonary vascular remodeling prior to birth in MAS.96 Although RDS primarily presents with signs of impaired alveolar ventilation, PPHN may complicate the course of RDS in late preterm and term neonates. The mortality and complication rates are increased for late preterm neonates who require ECMO for RDS and PPHN compared with term neonates.97 Although surfactant therapy is the primary modality for RDS, lack of improvement in oxygenation should lead to consideration of PPHN as a complication of this disease. The course of pneumonia and sepsis caused by group-B strep or gram-negative bacteria may be complicated by PPHN. Bacterial endotoxin can trigger the release of thromboxane, ET-1, and several cytokines including TNF-α that may cause pulmonary hypertension. In addition, sepsis is complicated by systemic hypotension and cardiac dysfunction, which contribute to right–left shunting and hypoxemia. Pulmonary hypoplasia from CDH and oligohydramnios is often associated with PPHN, which is more chronic from impaired pulmonary vascular growth. Lung hypoplasia decreases the capillary surface area, which leads to increased PVR. Many of these neonates show at least a transient response to vasodilator therapy, suggesting reactive vasoconstriction as a contributor of PPHN. In addition, neonates who die from severe lung hypoplasia also have increased medial and adventitial thickening of PAs.
Lethal Lung Diseases Associated with PPHN
Some neonates with PPHN who are resistant to medical therapy may have lung diseases, which are generally lethal. These include ACD and genetic defects in surfactant pathway.91–93 ACD, also referred to as misalignment of pulmonary veins, is diagnosed based on lung histology at lung biopsy or autopsy. ACD may occur as an isolated condition or in association with other congenital anomalies, with a preponderance of gastrointestinal, anorectal or renal malformations and CHD.91 Chromosomal deletions in the 16q24.1 to q24.2 regions were identified in sporadic cases of ACD/ misalignment of pulmonary veins in association with congenital anomalies. In addition, mutations in the FOXF1 gene have been associated with ACD in up to 40% of cases. Interestingly, FOXF1 gene maps to a small region of 16q24.1, suggesting that FOXF1 may be a candidate gene for ACD.91
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


