Chapter 64 Peripheral Vascular Anomalies, Malformations, and Vascular Tumors
Nonmalignant vascular anomalies can be functionally divided into two groups: proliferative vascular lesions and static vascular malformations. Unfortunately, this distinction is not universally appreciated, and diagnoses are often incorrect in the literature and clinical practice because of knowledge gaps and lack of clarity. Box 64-1A delineates a classification initially proposed by Mulliken and Glowaki,1 and Box 64-1B is an updated version published by the International Society for the Study of Vascular Anomalies (access at www.issva.org/). Functional classification helps guide management and prognosis. This chapter discusses peripheral (i.e., not central nervous system [CNS] or cardiac) vascular anomalies, including vascular tumors and syndromic vascular disorders, and offers new genetic information and insights into putative signaling pathways implicated in their development.
 Box 64-1A Functional Classification of Vascular Anomalies
Box 64-1A Functional Classification of Vascular Anomalies
 Box 64-1B Updated ISSVA Classification of Vascular Anomalies
Box 64-1B Updated ISSVA Classification of Vascular Anomalies
Vascular Tumors
Tufted angioma (± Kasabach-Merritt’s syndrome)
Kaposiform hemangioendothelioma (± Kasabach-Merritt’s syndrome)
Spindle cell hemangioendothelioma
Other rare hemangioendotheliomas (epithelioid, composite, retiform, polymorphous, Dabska tumor, lymphangioendotheliomatosis, etc.)
Acquired vascular tumors (pyogenic granuloma, targetoid hemangioma, glomeruloid hemangioma, microvenular hemangioma, etc.)
Vascular Malformations
Familial cutaneous and mucosal venous malformation (VMCM)
Fast-flow vascular malformations:
Complex-combined vascular malformations: CVM, CLM, LVM, CLVM, AVM-LM, CM-AVM
From Enjolras O, Wassef M, Chapot R: Color atlas of vascular tumors and vascular malformations, Cambridge, 2007, Cambridge University Press.
Proliferative Vascular Anomalies and Tumors
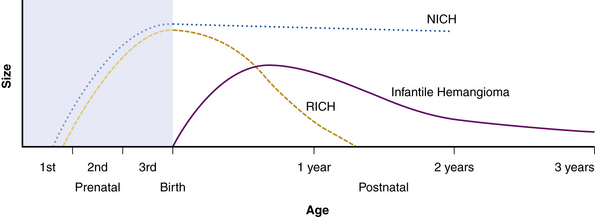
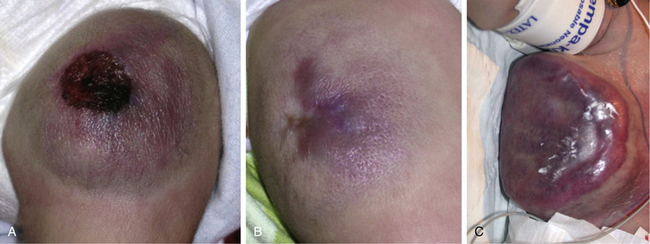
Hemangiomas are considered the most common tumors of childhood. They are benign growths of endothelial cells (ECs), with a unique natural history characterized by a rapid growth phase usually beginning in the first weeks of life and continuing until 9 to 12 months of age (Fig. 64-1). The majority of hemangiomas subsequently undergo spontaneous gradual (but extensive) involution. Histological correlation with the growth phase demonstrates involution and is characterized by increased connective tissue in the dermis and fat in the subcutaneous tissues.2 An important exception to this growth/regression pattern is the group of rapidly involuting congenital hemangiomas (RICH), which are generally present in full at birth (or even detected prenatally), and noninvoluting congenital hemangiomas (NICH), which do not change size postnatally.3 Growth curves for these hemangiomas are illustrated in Figure 64-2. A subset of patients with RICH may have high-flow lesions with prenatal or postnatal high-flow characteristics and/or transient coagulopathy4,5 (Fig. 64-3). Congenital nonprogressive hemangiomas have been shown by North et al. to be histologically and immunophenotypically distinct from classical hemangiomas of infancy and are speculated to have a differing pathogenesis.6 NICH-type lesions were found to have high flow clinically (as assessed by Doppler), and inferred histologically, in that small arteries were seen shunting into lobular vessels or abnormal veins.7 Another subtype of hemangiomas are those with minimal or arrested growth, presenting as areas of telangiectasia with peripheral bulkiness. In one series, the majority of this type of hemangioma was present on the lower extremities.8
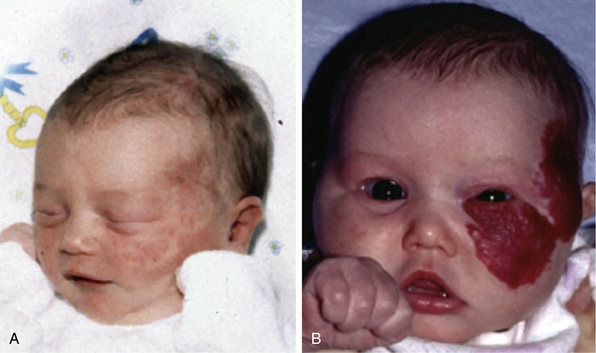
(From Mulliken J, Enjolras O: Congenital hemangiomas and infantile hemangioma: missing links. J Am Acad Dermatol 50:875–882, 2004; used with permission.)3
Typical hemangiomas are known to be most common in females, premature infants, and in the facial region. Results of the multicenter Hemangioma of Infancy Study of over 1000 children with hemangiomas showed an increased incidence in white non-Hispanic infants, multiple gestations, infants born to older mothers, and in association with placenta previa and/or preeclampsia.9 Other studies have shown (1) a threefold increased risk of hemangiomas in infants born to mothers who had transcervical chorionic villous sampling compared to amniocentesis (the incidence of hemangiomas in the amniocentesis group was equivalent to the incidence of hemangiomas in the general population)10,11 and (2) a correlation with placental anomalies with abnormal uteroplacental circulation.12,13 Waner et al. noted a nonrandom distribution of facial hemangiomas and found two patterns of growth: focal lesions (in 76.3% of the 205 patients assessed) and diffuse lesions (in 23.7%). The focal hemangiomas correlated to 22 sites of occurrence, all near lines of mesenchymal or mesenchymal-ectodermal embryonic fusion. The diffuse hemangiomas were in a segmental distribution and were specified as frontonasal (27%), maxillary (35%), or mandibular (38%). There was a threefold increased incidence of ulceration in patients with diffuse hemangiomas compared to that in patients with focal hemangiomas.14 Haggstrom et al. expanded the observation of nonrandom distribution, designating four primary segments (Seg1-Seg4) to correspond with cutaneous location15 (Fig. 64-4). Large hemangioma size, facial location, and/or segmental hemangiomas were more likely to require medical intervention.16 Segmental hemangiomas can be associated with a higher incidence of PHACE(S) syndrome, visceral hemangiomas, and underlying lumbosacral anomalies (e.g., occult spinal dysraphism, including lipomyelomeningocele with tethered cord).17–20PHACE(S) Association is an acronym for posterior fossa structural malformations, hemangiomas (segmental), arterial anomalies, cardiac defects, eye abnormalities, (and sternal and other midline deformities)21 (Fig. 64-5). A patient with a segmental hemangioma and one or more of these criteria has PHACES. In one series, approximately one third of patients with facial segmental hemangiomas were found to have PHACES, those at higher risk having large hemangiomas involving more than one anatomical segment, and in the frontonasal or frontotemporal distribution. Of those with PHACES, most (90%) had more than one extracutaneous finding (most commonly CNS arteriopathy or cardiac anomaly).22 Similarly, Oza et al. observed that patients with large facial segmental cutaneous (Seg1-Seg4) hemangiomas were especially at risk of CNS structural and cerebrovascular anomalies, those with Seg1 distribution hemangiomas had a higher incidence of ocular anomalies, and those with Seg3 distribution had airway, ventral, and cardiac anomalies. In this series, all patients with CNS structural anomalies had concomitant CNS arteriopathies. Also identified were supratentorial CNS anomalies (cortical dysgenesis and migration abnormalities). Arteriopathies are most commonly dysplastic vessels with an aberrant course involving the internal cerebral artery and its embryonic branches ipsilateral to the side of the cutaneous hemangioma.23 Hypoplasia, agenesis, or absence of normal arteries can also occur. In one review, some 20% of patients had arterial occlusions and stenoses.24 Progressive changes can lead to aneurysm formation.23
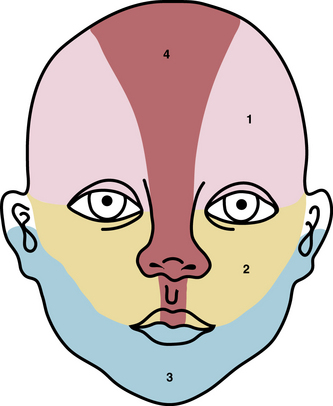
Figure 64-4 Distribution patterns of facial segmental hemangiomas.
(From Haggstrom AN, Lammer EJ, Schneider RA, et al: Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics 117:698–703, 2006.) Reproduced with permission, copyright by the AAP.
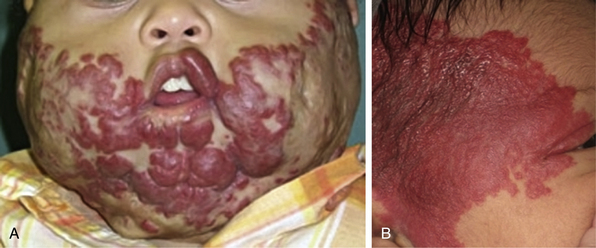
Kasabach-Merritt Phenomenon, Kaposiform Hemangioendothelioma, and Tufted Angioma
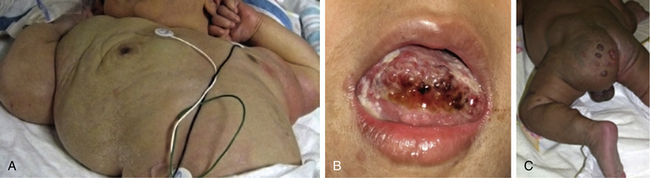
Trapping of platelets and other blood elements (Kasabach-Merritt phenomenon) has been known to occur in association with a subset of vascular anomalies since it was first described in 1940.25 This is an extremely important diagnosis because early detection and rapid evaluation and treatment (if clinically symptomatic) are essential. Kasabach-Merritt phenomenon is not associated with common hemangiomas of infancy, but with kaposiform hemangioendothelioma (KHE) or tufted angiomas.26,27 On examination, the lesion is often edematous, boggy, and ecchymotic (Fig. 64-6). Anatomical predilection is for the chest wall and shoulder, groin extending down the leg, retroperitoneum, or face. Gender distribution tends to be equal. Hematological features of Kasabach-Merritt phenomenon include thrombocytopenia, hypofibrinogenemia, elevated fibrin degradation products, and D-dimers. Radiological hallmarks of KHE are cutaneous thickening, diffuse enhancement with ill-defined margins, small feeding/draining vessels, stranding, and hemosiderin deposits. The histological features of KHE are spindled ECs resembling Kaposi sarcoma (but not associated with human immunodeficiency virus [HIV] infection), abnormal lymphatic-like vessels, microthrombi, hemosiderin, and decreased mast cells and pericytes (which are often seen in hemangiomas). There may be residual tumor after resolution of hematological abnormalities, and radiological studies often demonstrate persistent vascular tumors. Residua of KHE-associated tumors may be dormant vascular tumors rather than scars. Clinically as well as histologically, they differ considerably from involuted hemangiomas. A subset of patients with KHE do not have an associated coagulopathy.28 Treatment of KHE is not standardized but depends on the morbidity, location, and radiological features. Multimodal therapy may include steroids, chemotherapy (most commonly vincristine), interferon (IFN), antifibrinolytic agents, antiplatelet agents, and/or embolization. Diffuse intramuscular involvement often makes surgery not an option. Treatment with Rapamune (sirolimus) has been reported in one case2,29 and is currently being studied in a clinical trial (http://clinicaltrials.gov/ct2/show/NCT00975819?term=sirolimus&rank=82; see Table 64-4).
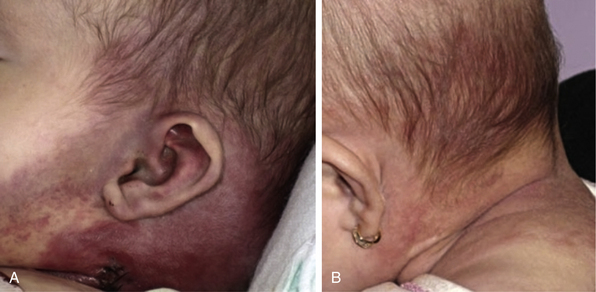
Tufted angioma, first described in the late 1980s, is a benign vascular tumor typified by tufts of capillaries in the dermis. The clinical appearance ranges from erythematous indurated annular nodules to plaques, with or without hypertrichosis (Fig. 64-7). They commonly occur on the trunk and extremities, and they may be associated with Kasabach-Merritt phenomenon. Chu et al. suggest that KHE and tufted angioma may represent a continuum; they report a case of transformation between both tumors within a single patient.30
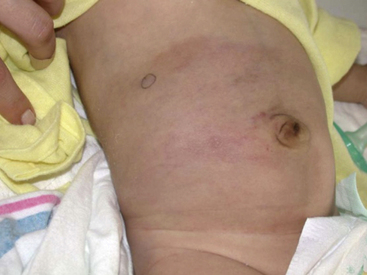
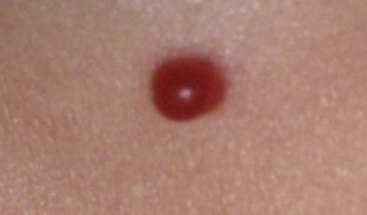
Pyogenic Granuloma
Pyogenic granuloma (also termed lobular capillary hemangioma) is an acquired vascular lesion of the skin and mucous membranes seen in pediatric patients (Fig. 64-8). The lesions have a cervicofacial propensity but can also be located on the trunk or extremities. The majority occur on the skin, and less frequently the mucous membranes (oral cavity and conjunctivae). These lesions are small and papular and tend to bleed. Treatment includes: (1) excision and linear closure, (2) shave excision, (3) cauterization, (4) cryotherapy, (5) carbon dioxide (CO2) or pulsed dye laser, or (6) sclerotherapy.31
Kaposi Sarcoma
Kaposi sarcoma is a neoplasm commonly but not exclusively seen in patients with acquired immunodeficiency syndrome (AIDS).32,33 It is an unusual vascular neoplasm originally described in 1872. The clinical appearance begins as violaceous macules that progress to plaques and papules and then nodules. Kaposi sarcoma is thought to be multifocal rather than metastatic, with multiple lesions occurring simultaneously at different anatomical locations. Histological features include spindle cells and ECs with rare mitotic figures. Evidence indicates that Kaposi sarcoma is monoclonal, although these data are conflicting. A novel human herpesvirus known as Kaposi sarcoma–associated herpesvirus (KSHV), or human herpesvirus type 8 (HHV8), has been identified in Kaposi sarcoma tissue, supporting a viral etiology. Growth factors and cytokines are also believed to be involved in Kaposi sarcoma development. Therapies directed against Kaposi sarcoma include antiviral agents, antiangiogenic drugs, and immunosuppressive agents. Recent studies show the effectiveness of antiretroviral therapy suppressing HIV/AIDS-associated Kaposi sarcoma growth.34
Vascular Malformations
Vascular malformations are present at birth and grow in parallel with the rate of growth of the child, with no propensity to spontaneous involution. They are due to developmental anomalies of the vasculature and may involve one or several types of vessels (arteries, veins, capillaries, or lymphatics). Vascular malformations are properly described according to the affected anomalous vascular channel. They can range from capillary malformations (commonly referred to as port-wine stains; Fig. 64-9) to large bulky growths that can distort the normal structures of the body and potentially lead to a high-output cardiac state (arterial malformations). Studies suggest that capillary malformations may be a result of abnormal innervation of discrete capillary beds causing chronic focal vascular ectasia.35,36 In one study, nerve density was significantly decreased in biopsies of capillary malformations, as compared to uninvolved skin.37
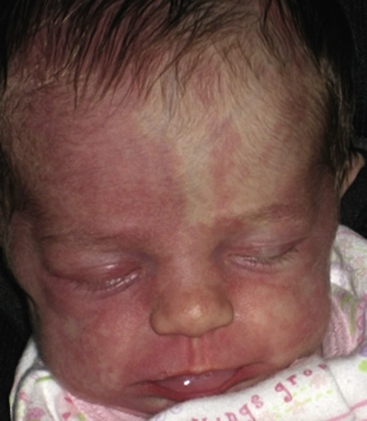
Figure 64-9 Capillary malformations.
Patient has facial capillary malformation and Sturge-Weber’s syndrome.
Lymphatic malformations may cause focal or generalized lymphedema, depending on the magnitude of aberrant lymphatics (Fig. 64-10). Abnormal growth of lymphatic circulation encompasses overdevelopment (in lymphangiodysplasias, lymphangiomas, and lymphangiomatosis), underdevelopment of lymphatic vasculature, or both. Disorders of the lymphatic circulation are common, diverse, and often devastating in their functional consequences. Clinical issues common to lymphatic anomalies reflect the tendency of these malformations to develop: (1) local and systemic infections/cellulitis (infectious and aseptic); (2) leakage (e.g., superficial blebs, chylous ascites, chylothorax, peritonitis, pleural effusions); (3) malabsorption syndromes with significant metabolic consequences; (4) craniofacial distortion interfering with swallowing, airway, or causing significant visceral dysfunction; (5) recurrences or complications after surgery; and (6) swelling of the affected anatomy, with functional limitation.38
Syndromic Vascular Anomalies
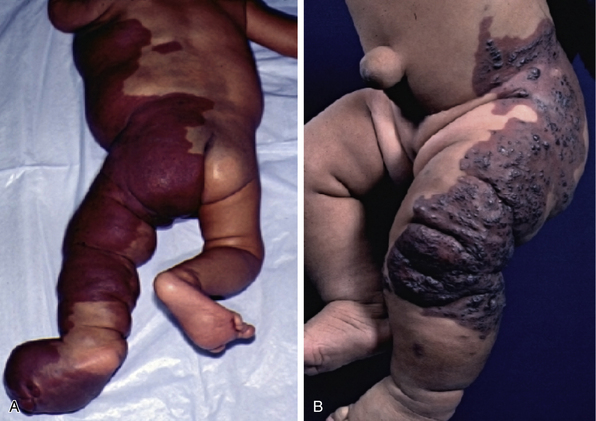
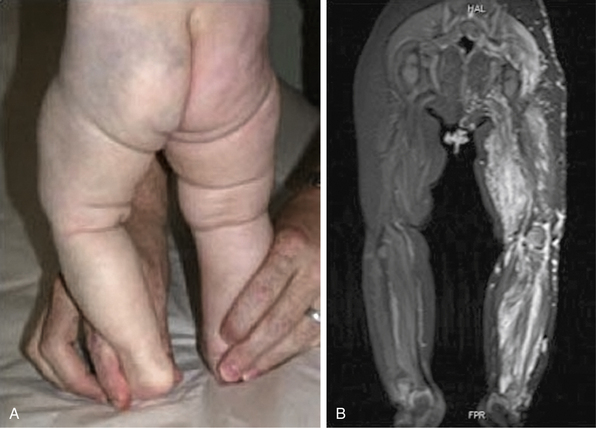
There is a spectrum of vascular malformations with dysregulated skeletal/adipose/soft-tissue growth (Table 64-1). Klippel-Trénaunay’s syndrome (capillary-lymphatic-venous malformation with ipsilateral limb enlargement or hypoplasia, venous varicosities or developmental anomalies, or both ) is one of the most common peripheral vascular malformation syndromes (Fig. 64-11). Males and females are affected in equal proportion, and the lower limb is the most frequent site of the anomaly. In severe cases, there may be an accompanying bleeding diathesis characterized by a normal to slightly decreased platelet count, decrease in fibrinogen, and increased D-dimers and fibrin degradation products39 (Fig. 64-12).
Table 64-1 Syndromic Vascular Anomalies and Genetic Information
| Name | Features | Omim |
|---|---|---|
| Blue rubber bleb nevus syndrome Bean syndrome | Multiple small soft venous malformations on skin, gastrointestinal tract, elsewhere | 112200 |
| CLOVES syndrome | Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi, skeletal/spinal anomalies | 612918 |
| Gorham’s syndrome Gorham-Stout’s syndrome Cystic angiomatosis of bone, diffuse disappearing bone disease | Lymphangiomatosis, bony destruction | 123880 |
| Klippel-Trénaunay’s syndrome | Capillary, venous, ± lymphatic malformation, hypertrophy of the related bones and soft tissues ± atretic deep venous system of affected extremity | 149000 |
| Maffucci’s syndrome (osteochondromatosis/ dyschondroplasia with vascular lesions) | Enchondromatosis and subcutaneous spindle cell hemangioendotheliomas, risk of chondrosarcoma, other malignancies including CNS | 166000 |
| Proteus’ syndrome | Gigantism (partial) of hands and feet, nevi, asymmetrical and disproportionate overgrowth, hemihypertrophy, macrocephaly, dysregulated adipose tissue, vascular malformations | 176920 |
| CM-AVM; CMC1 5q13-22 RASA-1 (RAS p21 protein activator 1) loss of function | Multifocal small macular CMs + AVM | 608354 |
| Venous malformations, multiple cutaneous and mucosal; VMCM 9p21 TIE2/TEK gain of function AD (most are sporadic) | Focal venous dilation with sparse vascular smooth muscle cells, cutaneous, mucosal, ± underlying areas | 600195 |
| Hennekam syndrome 18q21.32 CCBE1 Collagen and calcium-binding EGF domain–containing protein 1 | Intestinal lymphangiectasia, severe lymphedema, mental retardation | 235510 |
| Hypotrichosis-lymphedema-telangiectasia syndrome HLTS 20q13.33 SOX18 | Alopecia and/or areas of sparse hair, transparent skin, lymphedema, telangiectasia | 607823 |
| Lymphedema-distichiasis syndrome 16q24.3 AD or de novo FOXC2 loss of function | Limb edema and double rows of eyelashes (distichiasis) ± other associated anomalies including cardiac, renal, vascular, CNS gene mutation | 153400 |
| Milroy’s disease 5q35.3 AD, AR, or de novo FLT4 vascular endothelial growth factor receptor 3; VEGFR3 loss of function | Primary congenital hereditary lymphedema type Ia | 153100 |
| Lymphedema praecox Meige’s disease Late-onset lymphedema | Hereditary lymphedema type II Peripubertal onset | 153200 |
| Lymphangioleiomyomatosis LAM 16p13.3, 9q34 | Pulmonary (and extrapulmonary) lymphangiomyomatosis; female predominance, adult onset | 606690 |
| HHT Osler-Weber-Rendu AD Loss of function HHT type I 9q34.1 Endoglin (131195) Part of TGF-β receptor complex HHT type 2 12q11-q14 ALK1 Activin A receptor, type II-like kinase-1; ACVRLK1 cell surface receptor for TGF-β superfamily HHT type 3 5q31.3-q32 HHT type 4 7p14 Juvenile polyposis/HHT syndrome; JPHT 18q21.1 SMAD4 tumor suppressor; mutations affect TGF-β signaling | Cutaneous, mucosal and visceral telangiectasias and AVMs, epistaxis, and gastrointestinal bleeding, ± pulmonary AV fistulas, hepatic, CNS, spinal AVM HHT1: cerebral AVMs > pulmonary AVMs HHT2: hepatic AVMs more common | 187300 600376 601101 610655 175050 |
| Cutis marmorata telangiectatica congenita CMTC Macrocephaly-cutis marmorata telangiectatica congenita | Cutaneous reticulated mottling, telangiectasia, and phlebectasia, undergrowth or overgrowth of an involved extremity ± other anomalies | 219250 |
| Glomovenous malformation GVM AD 1p22-p21 Glomulin (601749) FKBP (FK506 binding proteins)-associated protein, 48-KD; FAP48 | Glomovenous malformations Cutaneous venous malformations with glomus cells surrounding distended vein-like channels | 138000 |
| PHACES Association | Posterior fossa brain malformations Segmental facial hemangiomas Arterial anomalies Cardiac anomalies Eye abnormalities Sternal or midline anomalies | 606519 |
| Bannayan-Riley-Ruvalcaba 10q23.31 PTEN Phosphatase and tensin homolog; tumor suppressor | Macrocephaly, multiple lipomas, vascular anomalies, pigmented macules of the penis | 153480 |
| Cowden’s syndrome 10q23.31 AD PTEN Phosphatase and tensin homolog; tumor suppressor PHTS | Macrocephaly, multiple hamartomas, cutaneous verrucous lesions, gingival/buccal papules, facial trichilemmomas, risk of breast/ thyroid/renal/endometrial malignancies, cerebelloparenchymal disorder VI (Lhermitte-Duclos’ disease) | 158350 |
Sturge-Weber’s syndrome includes a capillary malformation in the trigeminal distribution, intracranial angiomatosis and dysplasia, seizures, and glaucoma (see Fig. 64-9). Other examples of dysmorphic syndromes associated with vascular malformation are Turner’s and Noonan’s syndromes, Parkes Weber’s syndrome, hereditary hemorrhagic telangiectasia (HHT), blue rubber bleb nevus syndrome (Fig. 64-13), Maffucci’s syndrome, CLOVES syndrome (congenital lipomatous overgrowth, vascular malformations, epidermal nevi, spinal/skeletal anomalies or scoliosis), Proteus’s syndrome, Bannayan-Riley-Ruvalcaba’s syndrome, and Cowden’s syndrome (see Table 64-1). Syndromes noted for CNS vascular anomalies include von Hippel-Lindau, ataxia-telangiectasia, Sturge-Weber, and tuberous sclerosis; however, CNS and spinal arterial or venous anomalies are now known to occur in association with a number of vascular anomalies.23,40–45
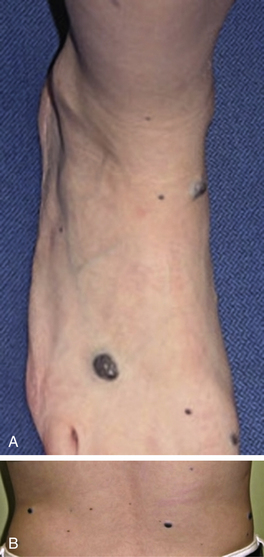
Dysmorphic syndromes associated with hemangiomas are predominantly associated with superficial segmental hemangiomas such as PHACES Association or sacral and/or genitourinary defects, associated with hemangiomas in the lumbar area.19,46
PTEN-Associated Hamartoma Syndromes
PTEN (phosphatase and tensin homolog protein) is a tumor suppressor gene. Patients with a PTEN mutation are susceptible to cancers and warrant early and regular screening. Some patients with vascular anomalies (arteriovenous, lymphatic, venous) have the PTEN mutation, such as those with Cowden’s and Bannayan-Riley-Ruvalcaba’s syndromes.47 A family history or presence of lipomas, thyroid disorders, tricholemmas, macrocephaly, and penile lentigines may point to a PTEN mutation. Consultation with a geneticist and family screening for mutations is indicated, and early screening for thyroid, breast, brain, gynecological, and other cancers should be initiated for all individuals with the PTEN mutation.47,48 Tan et al. recommend screening for PTEN mutations in patients with vascular malformations and the described findings and/or multiple vascular anomalies with a characteristic angiographic appearance, adipose-containing intramuscular lesions, and multiple intracranial developmental venous anomalies.40
Prenatal Diagnosis of Vascular Anomalies
With the availability of improved techniques in fetal ultrasound and magnetic resonance imaging (MRI), prenatally diagnosed vascular anomalies are becoming increasingly recognized. Most prenatally diagnosed vascular lesions are vascular malformations. Vascular lesions detected prenatally are generally identified by asymmetrical limbs and/or high-flow lesions (e.g., arteriovenous malformations [AVMs]or high-flow RICH-type lesions).49 If symptomatic in utero, such as high-flow vascular lesions compromising fetal hemodynamic status, prenatal therapy with maternal steroids or digoxin can be instituted. Maternal steroid therapy may be helpful in the management of fetal hemangiomas.50,51
Etiology of Hemangiomas and Vascular Malformations
Why do vascular anomalies occur? The simple answer is that they are due to many causes—mechanical, environmental, hormonal, and genetic—although no single etiology is thematic. Within the last several years, major research breakthroughs are unraveling potential etiological factors leading to formation of vascular anomalies, as detailed in excellent reviews.52–55
As subtypes of hemangiomas with segmental cutaneous distribution and associated visceral anomalies became evident, researchers speculated involvement of neural crest–derived cells, further supported by identification of neural crest cell markers (neurotrophin receptor p75) in proliferating hemangioma tissue.56 Several studies demonstrated markers for progenitor mesodermal stem cells (brachyury, GATA) or endothelial and hematopoietic cells (platelet endothelial adhesion molecule [PECAM]-1 [CD31]), intracellular adhesion molecule (ICAM)-3, bcl-2 gene expression, KDR+, CD133+, CD34+, endothelial precursor cells, lymphatic endothelial hyaluronan receptor-1, von Willebrand factor (vWF), and Snrk-1 in hemangioma tissue.57–60 Constitutive activation of the endothelial tie-2 receptor and vascular endothelial growth factor receptor (VEGFR)-2-related signaling pathways have been identified in human hemangiomas of infancy.52,54,61
Clonality of ECs was demonstrated,62,63 and the potential role of ECs in hemangioma development elucidated.64–66 Bischoff et al. isolated hemangioma-derived stem cells, which unlike other precursor cells, grew in vitro and differentiated in vivo into cells with properties of hemangiomas, including the eventual presence of adipocytes, as seen in involuting hemangiomas.2 Hemangiomas and placental vessels express common proteins including glucose transporter (GLUT)-1.67 This discovery is of diagnostic utility and spearheaded insights into placenta-based hypotheses. For example, Mihm and Nelson proposes a metastatic niche theory for hemangioma development, suggesting the placenta prepares hemangioma precursor cells that “home” to sites of hemangioma growth68 (Fig. 64-14). Proliferating hemangiomas have been shown to express VEGF-A as well as genes involved with nuclear factor (NF)-κB-related pathways.69,70
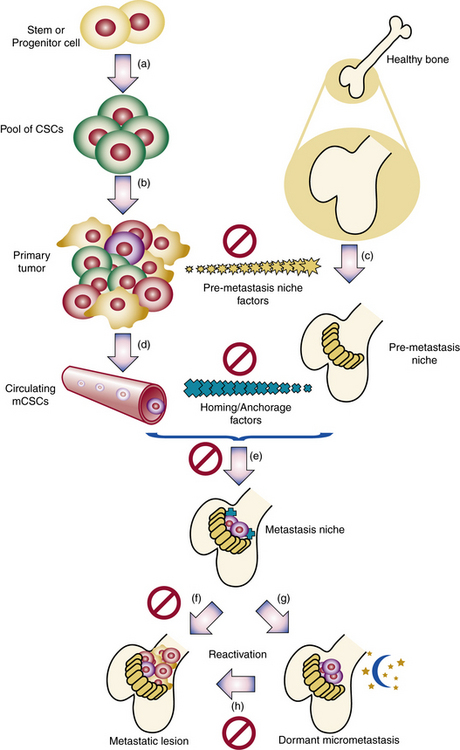
Figure 64-14 Metastatic niche theory of hemangioma development.
(From Mihm MC, Nelson JS: Hypothesis: the metastatic niche theory can elucidate infantile hemangioma development. J Cutan Pathol 37:83–87, 2010; used with permission.)
In addition, proapoptotic factors and appearance of adipocytes during the involution phase support a role for inflammation and immunoregulation in this process71 (Fig. 64-15). The vast majority of hemangiomas appear to be sporadic; however, familial cases harboring germline mutations of angiogenesis-related genes (VEGF2 and tumor endothelial matrix marker [TEM8]) have been identified.72 A secondary somatic event appears to be necessary for hemangioma development. Box 64-2 summarizes features of hemangioma endothelial cells.
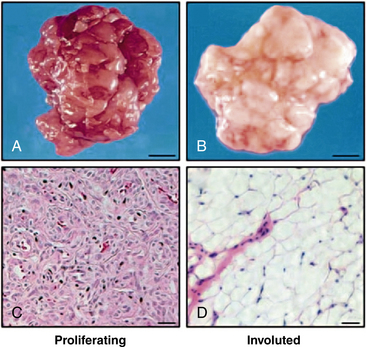
Figure 64-15 Adipocytes in hemangioma involution.
(From Yu Y, Fuhr, J, Boye, E, Gyorffy, S, Soker, S, Atalia, A, Mulliken, J, Bischoff. Mesenchymal Stem Cells and Adipogenesis in Hemangioma Involution. Stem Cells 2006;24:1605–1612.2 used with permission.)
 Box 64-2 Summary of Properties of Hemangioma Endothelial Cells*
Box 64-2 Summary of Properties of Hemangioma Endothelial Cells*
Express
Glut-1, vascular endothelial growth factor (VEGF) receptors, CD 31, CD34, VEGF-A, β-FGF, IGF-2, HIF-1α, Snrk-1, Tie-2, angiopoietin-2
Markers for progenitor mesodermal stem cell (brachyury, GATA)
Endothelial and hematopoietic cell markers (platelet endothelial adhesion molecule [PECAM]-1; CD31), intracellular adhesion molecule-2 (ICAM-3), bcl-2 gene expression, KDR+, CD133+, CD34+
Lymphatic endothelial hyaluronan receptor-1 (LYVE-1)
Angiotensin-converting enzyme (ACE) and angiotensin receptor 2 (ATR2)
Partially differentiated, resembling fetal endothelial cells in culture
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


