Chapter 67 Pediatric Surgery
Fluids, Electrolytes, and Nutrition
Fluid Requirements
Fluid requirements are calculated according to body weight (Table 67-1). During the first few days of life, the fluid recommendations are conservative; however, most neonates require 100 to 130 mL/kg/day for maintenance fluids by the fourth day of life. Neonates with conditions that are associated with excessive fluid losses (e.g., gastroschisis) can require as much as 1.5 times maintenance volume. The two best indicators of sufficient fluid intake are urine output and osmolarity. The minimum urine output in a newborn and young child is 1 to 2 mL/kg/day. Although adults can concentrate urine in the range of 1200 mOsm/kg, an infant responding to water deprivation is only able to concentrate urine to a maximum of 700 mOsm/kg. Clinically, this indicates that greater fluid intake and urine output are necessary to excrete the solute load presented to the kidney during normal metabolism. In general, the daily requirements for sodium and potassium are 2 to 4 and 1 to 2 mEq/kg, respectively. These requirements are usually met with a solution of 5% dextrose in 0.45% normal saline with 20 mEq KCl/liter at the calculated maintenance rate. Fluid losses from gastric drainage, ostomy output, or diarrhea should also be carefully assessed and replaced with an appropriate solution. Gastric losses should be replaced in equal volumes with 0.45% NS with 20 mEq KCl/liter. Diarrheal, pancreatic, and biliary losses are replaced with isotonic lactated Ringer’s solution. Acutely hypovolemic patients requiring rapid volume expansion should be treated with an IV bolus of 10 to 20 mL/kg body weight of whole blood, plasma, or 5% albumin. Transfusions of packed red blood cells are given in increments of 5 to 10 mL/kg.
Table 67-1 Daily Fluid Requirements for Neonates and Infants
| WEIGHT | VOLUME |
|---|---|
| Premature infants <2 kg | 140-150 mL/kg/day |
| Infants, 2-10 kg | 100 mL/kg/day for first 10 kg |
| Children, 10-20 kg | 1000 mL + 50 mL/kg/day for weight 10-20 kg |
| Children >20 kg | 1500 mL + 20 mL/kg/day for weight >20 kg |
Nutrition
Caloric Requirements
Energy requirements vary significantly from birth to childhood and also under different clinical conditions (Table 67-2). The parameter that is most indicative of sufficient delivery of calories in neonates is weight gain. Total daily caloric requirements and the expected daily weight gain decrease with age. Neonates have the highest energy requirements necessary to maintain growth. Almost 50% of the energy used in term infants younger than 2 weeks and 60% of energy intake in premature infants weighing less than 1200 g is devoted to growth. A general guideline for enteral caloric requirement for neonate is 120 calories/kg/day to achieve an ideal growth of 25 to 35 g/kg/day of weight gain (≈1% body weight gain/day). Most standard infant formulas, as well as breast milk, contain 20 calories/ounce. Formulas with higher caloric density are available for neonates who are unable to consume sufficient volumes to meet their caloric requirements and/or require fluid restriction. Breast milk or a protein hydrolysate formula (e.g., Pregestimil, Alimentum) should be used when beginning feedings in neonates with compromised gut functions (e.g., necrotizing enterocolitis [NEC]) or short gut caused by massive bowel resection. In general, continuous feedings are initiated for infants with a stressed gut and transition to bolus feedings is made later. Enteral feeding tolerance is carefully monitored by assessing for abdominal girth, gastric residuals, and stool or ostomy output.
Table 67-2 Average Caloric and Protein Requirement by Age
| AGE (yr) | CALORIES (kcal/kg/day) | PROTEIN (g/kg/day) |
|---|---|---|
| 0-1 | 90-124 | 2.0-3.5 |
| 1-7 | 75-90 | 2.0-2.5 |
| 7-12 | 60-75 | 2.0 |
| 12-18 | 30-60 | 1.5 |
| >18 | 25-30 | 1.0 |
Protein
The average intake of protein comprises approximately 15% of the total daily calories and ranges from 2 to 3.5 g/kg/day in infants. This protein requirement is reduced in half by age 12 and approaches adult requirement levels (1 g/kg/day) by 18 years of age (see Table 67-2). The provision of greater amounts of protein relative to nonprotein calories will result in rising blood urea nitrogen levels. The nonprotein calorie (carbohydrate plus fat calories)-to-protein calorie ratio (when expressed in grams of nitrogen) is therefore not less than 150 : 1. For infants receiving parenteral nutrition, the amount of protein provided is usually begun at 0.5 g/kg/day and advanced in daily increments of 0.5 g/kg/day to the target goal.
Neck Lesions
Cervical Lymphadenopathy
Cat scratch disease is a self-limiting infectious condition characterized by painful regional lymphadenopathy. Bartonella henselae, a gram-negative bacillus, is responsible for most cases. A history of exposure to cats is helpful, but not always present. Indirect immunofluorescent antibody testing has only moderate specificity and, therefore, the use of the polymerase chain reaction assay from a lymph node biopsy is more useful for diagnosis. There is no specific treatment for cat scratch disease because it is usually self-limited. A less common infectious cause for cervical lymphadenitis is nontuberculous mycobacterial infection.1 In general, the nodes are fluctuant, with a violaceous appearance of the overlying skin. The diagnosis is typically made by positive cultures for nontuberculous acid-fast bacilli, along with a tuberculin skin test. Surgical excision is usually indicated because most nontuberculous mycobacteria are resistant to conventional chemotherapy.
Cystic Hygroma
Aside from distorting adjacent normal structures, cystic hygromas are prone to infection and hemorrhage within the mass. Imaging studies, such as magnetic resonance imaging (MRI), can play a crucial role in preoperative planning. In general, complete surgical excision is the preferred treatment. However, this may be difficult because of the intimate involvement with surrounding vital structures. Resections are generally tedious, requiring careful isolation and ligation of lymphatic branches. Aggressive blunt and electrocautery dissections can lead to inadequate control of lymphatics, often resulting in recurrence and/or infection caused by accumulation of the lymphatic leak. Radical resection with sacrifice of vital structures is not advocated. Injection of sclerosing agents such as bleomycin or OK-432, derived from Streptococcus pyogenes, has been reported to be effective in the nonoperative management of cystic hygromas.2
Thyroglossal Duct Cyst
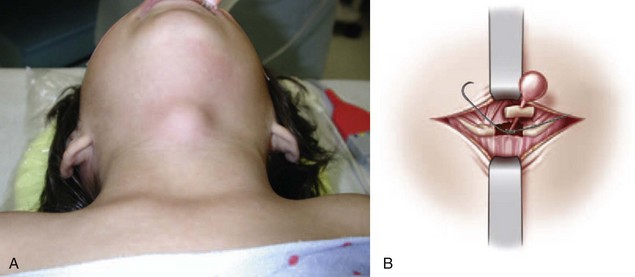
A thyroglossal duct cyst is a midline neck lesion that originates at the base of the tongue at foramen cecum and descends through the central portion of the hyoid bone. It is one of the most common midline neck lesions presenting in preschool-aged children (Fig. 67-1A). Although thyroglossal duct cysts may occur anywhere from the base of the tongue to the thyroid gland, most are found at or just below the hyoid bone. The standard operation for thyroglossal duct cysts has remained unchanged since it was described by Sistrunk in 1928. It involves complete excision of the cyst in continuity with its tract, the central portion of the hyoid bone, and the tissue above the hyoid bone, extending to the base of the tongue (see Fig. 67-1B). Failure to remove these tissues will result in a high risk for recurrence because multiple sinuses have been histologically identified in these locations. Embryologically, a thyroid diverticulum develops as a median endodermal thickening at the foramen cecum. As the embryo develops, the thyroid diverticulum descends in the neck and remains attached to the base of tongue by the thyroglossal duct. Also, as the thyroid gland descends to its normal pretracheal position, the ventral cartilages of the second and third branchial arches form the hyoid bone—hence, the intimate anatomic relationship of the thyroglossal duct remnant with the central portion of the hyoid bone. Normally, the thyroglossal duct regresses by the time the thyroid gland reaches its final position. When the elements of the duct persist despite complete thyroid descent, a thyroglossal duct cyst may develop. Failure of normal caudal migration of thyroid gland results in a lingual thyroid, in which no other thyroid tissue is present in the neck. Ultrasound or radionuclide imaging may provide useful information to identify the presence of a normal thyroid gland within the neck.
Extracorporeal Life Support
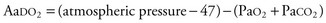
Physiologic Considerations
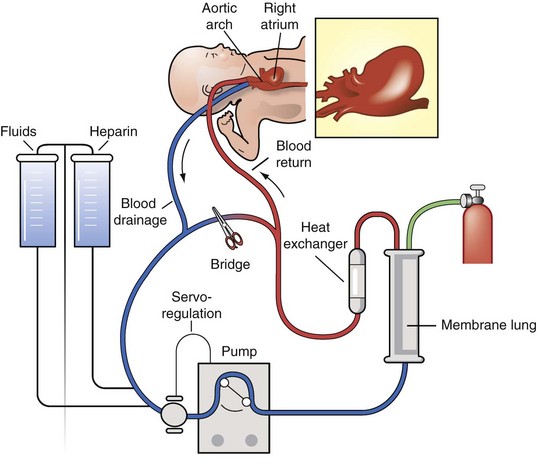
The basic concept of ECLS is to drain venous blood, remove carbon dioxide, add oxygen through the artificial membrane lung, and then return warmed blood back into the circulation. Venoarterial bypass provides cardiac and respiratory support, whereas venovenous bypass provides only respiratory support. Venoarterial bypass is used most commonly; the right internal jugular vein and common carotid artery are typically chosen for cannulation because of their vessel sizes, accessibility, and adequate collateral circulation. The ECLS circuit is composed of a silicone rubber bladder, which collapses when venous return is diminished, roller pump, membrane oxygenator, heat exchanger, tubing, and connectors. Venous blood from the right atrium drains through the venous cannula to the bladder and is pumped to the membrane oxygenator, where carbon dioxide is removed and oxygen is added (Fig. 67-2). The oxygenated blood then passes through the heat exchanger and is returned to the patient through the arterial cannula. The patient is systemically anticoagulated to prevent clotting of the ECLS circuit; hence, patients are at risk for bleeding complications. As such, hematocrit values, platelet counts, and fibrinogen levels must be closely monitored and maintained at acceptable ranges. Cranial ultrasound is performed for the first few days of ECLS to monitor for hemorrhage and then done on an as-needed basis. Extracorporeal flow is gradually weaned as native cardiac or pulmonary function improves. Indicators of lung recovery include an increasing PaO2, improved lung compliance, and clearing of the chest x-ray. Once the extracorporeal flow rate reaches minimal levels, the patient is trialed off bypass by temporarily clamping cannulas. If tolerated, the patient is taken off ECLS support on moderate conventional ventilatory settings.
Congenital Diaphragmatic Hernia
Diagnosis
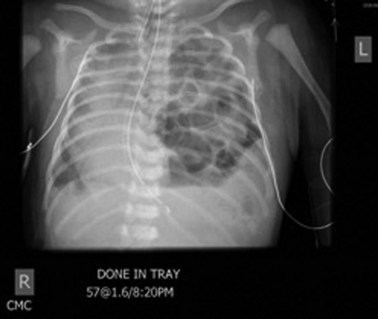
At birth, CDH infants demonstrate symptoms of respiratory distress with a classic chest radiographic appearance of multiple bowel loops in the thoracic cavity, along with mediastinal shift (Fig. 67-3). An orogastric tube may appear to coil in the chest. The differential diagnosis of CDH includes congenital cystic adenomatoid malformation, bronchogenic cyst, diaphragmatic eventration, and cystic teratoma. In Morgagni’s hernia, the diagnosis is often delayed until childhood because most infants are asymptomatic. Chest radiographs may reveal an air-fluid level immediately posterior to the sternum. Usually, the infant does well for several hours after delivery during the so-called honeymoon period and then begins to demonstrate worsening respiratory function. Therapeutic interventions are targeted to reduce PPHN. In approximately 10% to 20% of cases, CDH is diagnosed beyond the first 24 hours of life, at which time infants present with various symptoms of feeding difficulties, respiratory distress, and pneumonia.
Treatment
Surgical Repair
It has been well established that CDH repair should be delayed for 2 to 4 days until cardiopulmonary stabilization has occurred. The preferred operative approach for a posterolateral CDH is through a subcostal abdominal incision. The viscera are reduced into the abdominal cavity and the posterolateral defect in the diaphragm is closed using interrupted nonabsorbable sutures. When present (10% to 15% of cases), a hernia sac should be excised. Typically, the hernia defect is large, with only a small anteromedial leaflet of diaphragmatic tissue present. A number of reconstructive techniques and materials are available for the repair of large hernia defects. The surgical technique of abdominal or thoracic muscle flaps can be considered, but the use of prosthetic material (e.g., Gore-Tex, W.L. Gore, Elkton, Md) has become more widespread. The advantages of a prosthetic patch are shorter operative time and a tension-free repair. However, the major potential problems with prosthetic patches are the risks of infection and recurrence of the hernia. Recently, the use of regenerative extracellular matrix biomaterials has garnered significant interest as an ideal biodegradable patch to repair diaphragmatic hernia defects. At times, the abdominal cavity may be too small to accommodate the reduced viscera from the thoracic cavity. A temporary abdominal silo may be considered, but allowing for an incisional hernia with skin-only closure until the definitive fascia closure can be performed is an alternative surgical option. The timing of CDH repair relative to ECLS remains controversial. A recent CDH study group report has suggested that CDH repair after ECLS therapy is associated with improved survival when compared with repair while on ECLS.3
Bronchopulmonary Malformations
Bronchogenic Cyst
Bronchogenic cyst is the most common cystic lesion of the mediastinum. The cyst wall consists of fibroelastic tissue, smooth muscle, and cartilage, whereas the cyst itself is lined with respiratory tract epithelia (ciliated columnar cells). It can also contain mucus-producing cuboidal cells, which contribute to enlargement of the cyst with mucus. They may occur anywhere along the tracheobronchial tree but are usually found around the carina and right hilum. Less frequently, they present in the neck, lung, pleura, pericardium, or below the diaphragm. When the cysts are large, they can compress surrounding vital structures, including the airway. Infants are particularly at risk because of their narrow, easily compressible airway. Bronchogenic cysts can also cause dysphagia, pneumothorax, cough, and hemoptysis or become infected, which is how older children present. Often picked up on postnatal chest x-ray, the diagnosis is confirmed by CT as a spherical nonenhancing mass. It is fluid- or mucus-filled, although an air-fluid level is apparent if the cyst communicates with the airway. Cysts within the pulmonary parenchyma typically communicate with a bronchus, whereas those in the mediastinum usually do not. Bronchogenic cysts are routinely resected even if asymptomatic, although recently there have been debates on observation alone for this condition.4 Rare cases of malignant transformation have been reported. Resection is performed by video-assisted thoracic surgery (VATS) or thoracotomy.
Congenital Pulmonary Airway Malformation
CPAMs have been controversially described as hamartomatous lesions in which a multicystic mass replaces normal lung tissue. They are connected to the tracheobronchial tree and its blood supply is pulmonary. Although they are usually unilateral and unilobar, they can present in the immediate perinatal period with life-threatening respiratory distress. If asymptomatic during this time, infants and older children can go on to present with fevers, persistent cough, and recurrent pneumonia. CPAMs can undergo malignant transformation; rhabdomyosarcoma has been reported. They are classified based on their appearance on imaging and confirmation is made by pathologic examination. According to the Stocker classification, type I lesions account for almost 75% of all cases and consist of a small number of large, 2- to 10-cm cysts that can compress normal lung parenchyma. Type II lesions have numerous cysts, usually measuring less than 1 cm in diameter. Type III lesions are rare and appear to be only a few millimeters in diameter.5 However, they are associated with mediastinal shift, hydrops, and a poor prognosis.
Ultrasound and, less commonly, MRI, are used to locate the lesion, characterize its appearance, determine its blood supply and venous drainage, and evaluate whether there is any displacement of other thoracic structures. Also, when there is any uncertainty, prenatal MRI has been used to differentiate CPAM from other congenital thoracic abnormalities. Postnatally, a chest radiograph is usually diagnostic, revealing a mass with a possible mediastinal shift and air-fluid levels, but a chest CT is frequently obtained for confirmation. When fetal distress occurs in utero, options include fetal thoracotomy and thoracoamniotic shunting (if the fetus is <32 weeks). Most fetuses with a prenatally diagnosed CPAM experience partial regression in the third trimester and can be treated with expectant management. They can then undergo postnatal resection at 5 to 8 weeks of life.4 Postnatal management of the symptomatic patient necessitates resection via a thoracotomy or VATS. The treatment of asymptomatic patients is more controversial but it is generally agreed that they should be resected, given the risk of infection and malignancy.
Pulmonary Sequestration
Bronchopulmonary sequestrations (BPSs) are nonfunctional nests of microcystic pulmonary tissue that have no connection to the tracheobronchial tree but are fed by an aberrant systemic artery. There are two types, intralobar (IL) and extralobar (EL); the former is contained within normal lung parenchyma and the latter is separate and encased by its own pleura.4 ELSs occur predominantly in males and, in 40% of cases, other congenital anomalies, such as posterolateral diaphragmatic hernia, pectus excavatum and carinatum, and enteric duplication cysts, can be found.
Lacking a communication to the airway, sequestrations do not form enlarged cysts or cause spontaneous pneumothoraces. They can, however, infarct, become infected, and cause hemoptysis. It has been reported that ELSs can undergo torsion as well. Because of their aberrant systemic vascular supply (Fig. 67-4), BPSs can result in significant left-to-right shunting in infants, who are then susceptible to high-output cardiac failure.6 For an initial evaluation, Doppler ultrasound may reveal a systemic arterial supply from the infradiaphragmatic or thoracic aorta. The lesion itself may appear solid, but can also be cystic. CT or MRI can aid in further defining the vascular anatomy. Accounting for 75% of all BPSs, intralobar sequestrations (ILSs) are found within the medial or posterior segments of the lower lobes, more on the left side. Most ELSs are found posteromedially in the left lower chest but can occur within or below the diaphragm. Air within an ILS usually signifies infection, whereas the same finding in an ELS suggests the presence of a fistulous connection with the esophagus.4
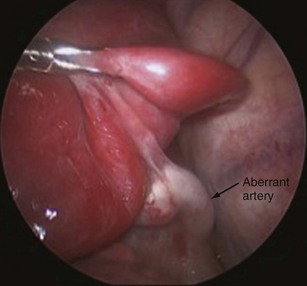
If a BPS is identified on prenatal ultrasound, the fetus is followed with serial ultrasound, looking for mass enlargement and the development of complications such as hydrops, pleural effusion, or polyhydramnios.6 BPS has been reported to spontaneously regress. In fact, it is estimated that 68% of BPSs undergo spontaneous regression before birth as they become isodense with the surrounding lung. The involution may occur as the lesion outgrows its blood supply. Because of the risk for infection and bleeding, ILSs are usually resected by segmentectomy or lobectomy. ELSs are usually asymptomatic and, because there is generally no tracheobronchial communication, the risk of infection is low. As such, many of these lesions can be observed.
Congenital Lobar Emphysema
CLE describes a progressively distended, hyperlucent lobe caused by abnormal bronchopulmonary development. Air trapping in the emphysematous lobes occurs with intrinsic or extrinsic obstruction, which includes endobronchial obstruction from mucosal proliferation and extrinsic compression from vascular anomalies. In over 90% of cases, it involves the left upper or right middle lobe. CLE is rarely diagnosed prenatally, but its prevalence is 1 in every 20,000 to 30,000 deliveries.4 It tends to present in the first few days of life and as late as 6 months after birth. On ultrasound, CLE appears as an echogenic homogenous lung mass. When CLE is discovered in an asymptomatic patient, observation is recommended because these lesions have a tendency to regress. A chest radiograph is customarily diagnostic because it reveals overdistention of the involved lobe. Importantly, the lucency should not be mistaken for a pneumothorax, and positive-pressure ventilation should be used with caution because of the propensity of these patients to undergo auto-PEEP (positive end-expiratory pressure; auto-PEEP is defined as the end-expiratory intrapulmonary pressure that develops as a result of dynamic airflow resistance during mechanical ventilation). When the CLE progresses to the point that it causes mediastinal shift and worsening symptoms, an open thoracotomy with lobectomy is indicated.
Alimentary Tract
Esophageal Atresia and Tracheoesophageal Fistula
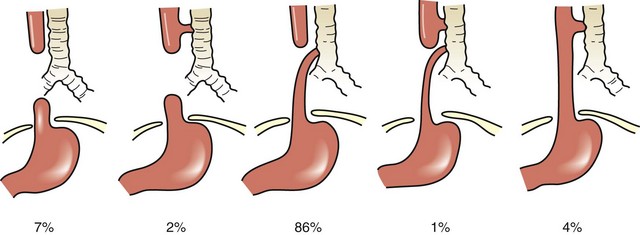
Esophageal atresia is a congenital condition of esophageal discontinuity that results in proximal esophageal obstruction. A tracheoesophageal fistula (TEF) is an abnormal fistula communication between the esophagus and trachea. Esophageal atresia and TEF can occur alone or in combination. The incidence of this anomaly is 1 in 1500 to 3000 live births, with a slight male predominance. Approximately one third of infants with esophageal atresia or TEF have a low birth weight, and 60% to 70% have associated anomalies. During the fourth week of gestation, the esophagotracheal diverticulum of the foregut fails to divide completely to form the esophagus and trachea. In 10% of patients, there is a nonrandom, nonhereditary association of anomalies referred to by the acronym VATER (vertebral, anorectal, tracheal, esophageal, renal or radial limb); an alternative acronym is VACTERL (vertebral, anorectal, cardiac, tracheal, esophageal, renal, and limb). Five anatomic variants of esophageal atresia are depicted in Figure 67-5. In the most common type (C lesion) of esophageal atresia with TEF, the proximal blind pouch ends approximately the distance of one to two vertebral bodies from the distal TEF. The distal TEF is typically located 1 cm above the carina in the membranous portion of the trachea.
Clinical Presentation and Diagnosis
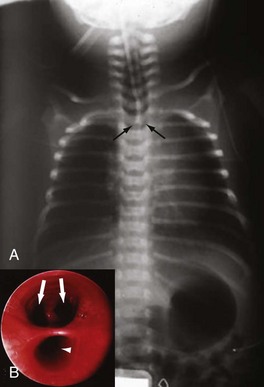
The diagnosis of esophageal atresia is considered in an infant with excessive salivation along with coughing or choking during the first oral feeding. A maternal history of polyhydramnios is often present, more often in isolated proximal atresia (86%). In an infant with esophageal atresia and TEF, acute gastric distention may occur as a result of air entering the distal esophagus and stomach with each inspired breath. Reflux of gastric contents into the distal esophagus will traverse the TEF and spill into the trachea, resulting in cough, tachypnea, apnea, and/or cyanosis. The presentation of isolated TEF without esophageal atresia may be more subtle, often beyond the newborn period. In general, these infants have choking and coughing associated with oral feeding. The inability to pass a nasogastric tube into the stomach of the neonate is a cardinal feature for the diagnosis of esophageal atresia. If gas is present in the GI tract below the diaphragm, an associated TEF is confirmed (Fig. 67-6A). Conversely, the inability to pass a nasogastric tube in an infant with absent radiographic evidence of GI gas is almost diagnostic of an isolated esophageal atresia. The use of isotonic contrast by mouth to demonstrate the presence and/or level of the proximal esophageal atresia is strongly discouraged because of risk for aspiration. The diagnostic evaluation includes screening for other associated anomalies. Echocardiography and renal ultrasound are performed to evaluate for congenital heart defects (including aortic arch anomaly) and genitourinary malformations.
Treatment
Preoperative Management
Initial treatment includes decompression of the proximal esophageal pouch with a sump tube (e.g., Replogle tube) placed on continuous suction. The infant is positioned in an upright prone position to minimize GER and prevent aspiration. Broad-spectrum IV antibiotic coverage should be started empirically. Routine endotracheal intubation is avoided because positive-pressure ventilation may be inadequate to inflate the lungs because air is directed into the TEF through the path of least resistance. Ventilation may be compounded further by the resultant gastric distention. Gastrostomy to decompress the distended stomach should be avoided because it may abruptly worsen the ability to ventilate the patient. In these circumstances, manipulation of the endotracheal tube to be positioned distal to the TEF (e.g., right mainstem intubation) may minimize the leak and permit adequate ventilation. Furthermore, placement of an occlusive balloon catheter (Fogarty) into the fistula through a bronchoscope may be useful, but this maneuver can be risky. As a last resort, emergent thoracotomy with ligation of the fistula alone may be required. A preoperative chest radiograph and echocardiogram provide sufficient information to determine the side of the aortic arch. A right thoracotomy is performed for the operative repair in patients with a normal left-sided aortic arch. However, for infants with a right-sided arch, a left thoracotomy would be preferred. A higher incidence of aortic arch anomalies (e.g., vascular rings) and postoperative complications has been reported with right-sided aortic arch.7
Surgical Management
The typical surgical approach for the most common esophageal atresia with TEF involves an open thoracotomy, with an extrapleural approach through the fourth intercostal space. Bronchoscopy is routinely performed to determine the relative site of the fistula and exclude the presence of a second fistula (see Fig. 67-6B). After extrapleural dissection to expose the posterior mediastinum, the azygos vein is divided to reveal the underlying TEF. The TEF is dissected circumferentially and its attachment to the membranous portion of trachea is taken down. The tracheal opening is approximated using interrupted nonabsorbable sutures. The proximal esophageal pouch is then mobilized as high as possible to afford a tension-free esophageal anastomosis. The blood supply to the upper esophageal pouch is generally robust and is based on arteries derived from the thyrocervical trunk. However, the blood supply to the lower esophagus is more tenuous and segmental, originating from intercostal vessels. As such, significant mobilization of the lower esophagus is avoided to prevent ischemia at the esophageal anastomosis. The anastomosis is performed using a single- or double-layer technique (Fig. 67-7A). The rates of anastomotic leak are slightly higher with the single-layer anastomosis, whereas the rates of esophageal stricture are higher with the double-layer technique.
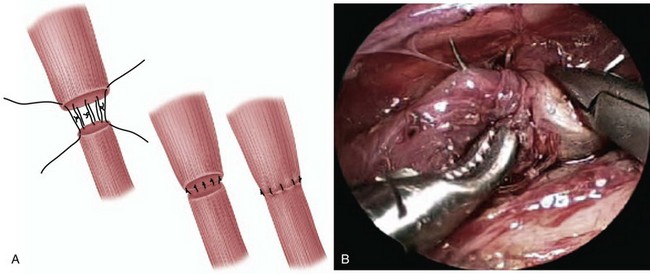
In case of a long gap between the two ends of the esophagus, there are several options. The first option is to suture the divided end of the distal esophagus to the prevertebral fascia, mark its location with a metal clip, and close the thoracotomy. Over a 2- to 3-month period, the proximal esophageal pouch may grow so that a subsequent primary esophageal anastomosis may be possible. Second, a circular or spiral esophagomyotomy of the upper pouch may be performed to gain esophageal length and facilitate a primary anastomosis. Another technique involves the placement of traction sutures through the proximal and distal ends of the esophagus, brought out through the chest. These sutures are then progressively tightened and a primary esophageal anastomosis is performed after several days.8 Alternatively, a cervical esophagostomy may be constructed and a formal esophageal replacement performed at a later date.
In patients with pure esophageal atresia, primary anastomosis in the newborn period is not feasible because of a long gap between the two esophageal ends. Initially, a cervical esophagostomy for drainage of oral secretions and gastrostomy for enteral feeding access are performed. An esophageal replacement using the stomach, small intestine, or colon is then carried out at approximately 1 year of age. In some cases, the two ends of the esophagus may spontaneously grow so that a primary anastomosis may be accomplished by 4 months of age. The swallowing of saliva may actually promote elongation of the upper pouch, and an esophagostomy is therefore avoided. In patients with pure TEF, without esophageal atresia, the site of the TEF is usually in the region of the thoracic inlet. In this case, the surgical approach is through a cervical incision. At the time of surgical repair, it is often helpful to perform rigid bronchoscopy and cannulate the TEF with a guidewire to facilitate its identification. Recently, thoracoscopic repair has been described by several pediatric surgical centers (see Fig. 67-7B).9
Gastroesophageal Reflux
Clinical Presentation
Pathologic symptoms of GER vary considerably, depending on the age of the patient and underlying associated medical conditions. Although vomiting is a common symptom, failure to thrive (FTT) as a result of caloric deprivation is one of the most critical complications of persistent GER in infants and children. Aspiration of gastric contents can also result in recurrent bronchitis or pneumonia, presenting initially with a chronic cough or wheezing. Reflux may stimulate vagal reflexes, producing laryngospasm or bronchospasm and leading to an asthma-like clinical picture.10 The significant airway spasm caused by reflux can result in apnea or choking spells, and may contribute to near-miss sudden infant death syndrome (SIDS). Irritability and crying in infants may also represent pain because of esophagitis induced by chronic reflux. Chronic acid insult to the lower esophagus can progress to the formation of stricture from chronic scarring and produce obstructive symptoms. Although rare in pediatric patients, chronic progressive esophagitis can lead to metaplastic replacement of normal lower esophageal squamous mucosa by columnar epithelium. This condition, known as Barrett’s esophagus, requires close surveillance to detect the progression of premalignant dysplastic changes.
Many children referred for antireflux surgery are neurologically impaired, usually secondary to such factors as metabolic conditions, head trauma, or birth asphyxia. Thus, most of these patients require permanent feeding access in the form of a gastrostomy tube, so antireflux surgery is often considered at the time of the gastrostomy placement, especially in patients who are unable to protect their airway reliably or who already have significant vomiting associated with intragastric tube feeding. However, the concept of prophylactic fundoplication at the time of gastrostomy in neurologically impaired children remains controversial. These patients are also at high risk for delayed gastric emptying because of upper gastroduodenal dysmotility. However, fundoplication alone may result in enhanced gastric emptying function.11
Hypertrophic Pyloric Stenosis
Treatment: Surgical Management
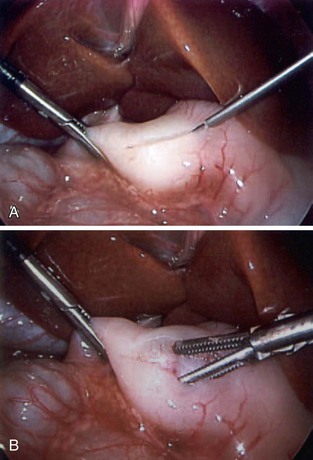
The treatment of HPS is pyloromyotomy, consisting of incising through thickened pyloric musculature while preserving the underlying mucosa. This can be performed through a right upper quadrant or periumbilical incision. Recently, laparoscopic pyloromyotomy (Fig. 67-8) has gained popularity because of better cosmesis, with similar outcomes to those of the open technique.12 Before surgery, it is important that the infant be fully rehydrated with IV fluids to establish an adequate urine output and correct electrolyte disturbances such as metabolic alkalosis. Because the infant with underlying metabolic alkalosis will compensate with respiratory acidosis, postoperative apnea may occur. Thus, the serum HCO3− level needs to be normalized before surgery. Postoperatively, infants are usually allowed to resume enteral feedings. Vomiting after surgery occurs frequently but is generally self-limited. Potential complications include incomplete myotomy, mucosal perforation, and wound infection.
Duodenal Atresia
Clinical Presentation and Diagnosis
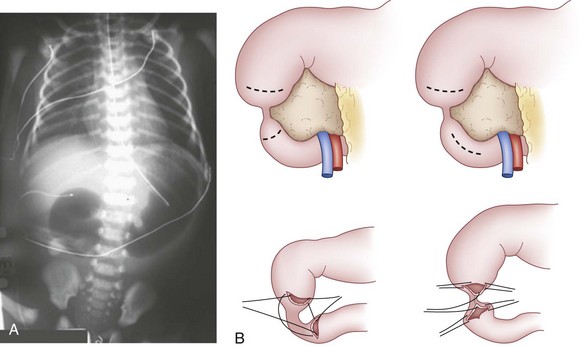
Infants with duodenal obstruction are generally first detected during a prenatal ultrasound evaluation. Immediately after birth, a plain abdominal radiograph shows a typical double-bubble sign, if obtained before orogastric tube decompression of swallowed gastric air (Fig. 67-9A). If distal air is present, an upper GI contrast study should be obtained rapidly, not only to confirm the diagnosis of duodenal stenosis or atresia but also to exclude midgut volvulus, which would constitute a surgical emergency.
Treatment: Surgical Management
The management is by surgical bypass of the duodenal obstruction as a side-to-side or proximal transverse to distal longitudinal (diamond-shaped) duodenoduodenostomy (see Fig. 67-9B). At the time of anastomosis, a concomitant distal intestinal atresia should be ruled out by injecting saline into a distal limb using a soft red rubber catheter. When the proximal duodenum is markedly dilated, a tapering duodenoplasty with staples or sutures should be considered to reduce the duodenal caliber, which may improve postoperative gastric emptying. In patients with a duodenal mucosal web, the web is excised transduodenally. Caution must be exercised to preserve the ampulla during the web excision.
Jejunoileal Atresia
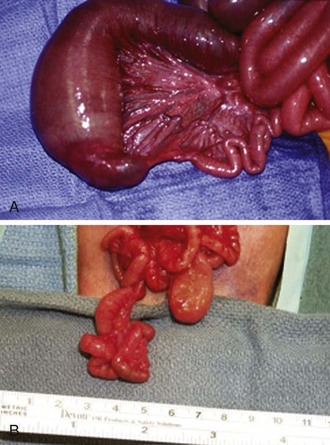
Jejunoileal atresia is the most common GI atresia; it occurs in approximately 1 in 2000 live births. It is thought to occur as a result of an intrauterine mesenteric vascular occlusion. Atresias occur slightly more frequently in the jejunum than in the ileum. Jejunoileal atresias are classified as type I, a mucosal web or diaphragm (Fig. 67-10A), type II, with an atretic cord between two blind ends of bowel with intact mesentery, type IIIa, a complete separation of the blind ends of the bowel by a V-shaped mesenteric gap, and type IIIb, an apple peel or Christmas tree deformity with a large mesenteric gap (see Fig. 67-10B), in which the distal bowel receives a retrograde blood supply from the ileocolic or right colic artery. This tenuous blood supply has implications for reanastomosis and the potential for ischemic necrosis caused by an antenatal volvulus. Thus, many of these infants with this type of atresia are born with reduced intestinal length. Finally, in type IV, there are multiple atresias, with a string of sausage appearance.