Pediatric Pulmonary Hypertension
Dunbar Ivy
Pulmonary hypertension (PH) is an important cause of morbidity and mortality in pediatric patients (1,2,3). Advances in understanding the pathobiology of pulmonary arterial hypertension (PAH) have led to novel therapies; however, there remains no cure for many forms of PH, such as idiopathic pulmonary arterial hypertension (IPAH) (4). Evaluation and treatment for known causes of PH are crucial as the best strategy includes treatment of any underlying disorders. The selection of appropriate therapies for PH is complex and must be carefully chosen according to the etiology and determination of pulmonary vasoreactivity at cardiac catheterization.
Definition and Classification
Pulmonary hypertension is defined as a mean pulmonary artery pressure (PAP) greater than 25 mm Hg at cardiac catheterization in a resting state and has many causes (5,6). PAH describes a subpopulation of patients with PH characterized hemodynamically by the presence of precapillary PH including an end-expiratory pulmonary artery wedge pressure (PAWP) ≤15 mm Hg and a pulmonary vascular resistance >3 Wood units (7) (Table 66.1).
PH is no longer classified as primary or secondary. A revision of the WHO classification, including most of the forms of PH encountered in children, was proposed at the World Symposium of Pulmonary Hypertension (WSPH) Dana Point meeting in 2008 (7) and updated at the WSPH in Nice in 2013 (Table 66.2) (1); however, this classification was not designed specifically for children. The causes of PH have been grouped into five categories; each category shares similar features, such as treatment and pathobiology, but there is overlap between groups. Group 1 includes patients with PAH. PAH may be idiopathic (IPAH) or heritable (HPAH) with no underlying cause, or associated with a specific disease (associated PAH). IPAH is a diagnosis of exclusion and thus is defined as PAH unexplained by other causes. Thus, an extensive evaluation to rule out secondary causes must be performed before a patient can be diagnosed with IPAH. Mutations of the bone morphogenetic protein receptor
type 2 (BMPR2) gene are the most common genetic abnormality known to cause HPAH (8). Most studies in children have focused on PAH associated with congenital heart disease (CHD) (Tables 66.3 and 66.4) and IPAH (9,10,11). Patients with PAH and CHD are a heterogeneous population, with a range of vascular disease: some patients have a right-to-left central shunt from severe pulmonary vascular obstructive disease (Eisenmenger syndrome), others have a residual shunt and severe PAH but are not cyanotic, some patients with a small shunt may have severe PAH and are more similar to IPAH, and finally it is well known that PAH may persist and progress despite corrective cardiac surgery. Persistent pulmonary hypertension of the newborn (PPHN) is the most common cause of PH in the neonatal period, and is most often reversible (12,13). PAH associated with pulmonary veno-occlusive disease (PVOD) (14,15,16) or pulmonary capillary hemangiomatosis (PCH) (16,17,18) is also categorized in group 1. Mutations in EIF2AK4, a gene which regulates angiogenesis in response to cellular stress, have been identified both in patients with PVOD and PCH suggesting a common pathogenesis. PH associated with left heart disease (“venous hypertension”) is defined as group 2 PH. In these disorders, the main cause of PH is an elevation of left heart pressure due to impaired relaxation (diastolic dysfunction) or an anatomic lesion that leads to high left atrial pressure (such as mitral stenosis). Abnormalities of left heart inflow or outflow, such as cor triatriatum or subaortic stenosis, were recently added to the Nice classification in 2013. The initial treatment of most of these disorders generally is treatment of the anatomic obstruction as pulmonary vasodilators may lead to pulmonary edema. Of note, patients with a cavopulmonary anastomosis are not included in the Nice classification. Group 3 PH, or PH associated with lung disease or hypoxemia, is separated from group 1 as treatment of the lung disorder may result in normalization of PAP. PH associated with chronic lung diseases, such as bronchopulmonary dysplasia (BPD) (19,20,21) or congenital diaphragmatic hernia (CDH) (22,23,24,25) is increasingly recognized as an important cause of PH in the infant and child. The Nice classification includes a list of lung development abnormalities associated with PH (Table 66.5) (1). Group 4 PH, or PH associated with chronic thromboembolic disease, is a rare cause of PH in children, but is still important to recognize and treat as therapy may be curative (26). Group 5 consists of disorders with multifactorial etiology, such as SCD (27,28) or segmental PH as seen in pulmonary atresia with ventricular septal defect (VSD).
type 2 (BMPR2) gene are the most common genetic abnormality known to cause HPAH (8). Most studies in children have focused on PAH associated with congenital heart disease (CHD) (Tables 66.3 and 66.4) and IPAH (9,10,11). Patients with PAH and CHD are a heterogeneous population, with a range of vascular disease: some patients have a right-to-left central shunt from severe pulmonary vascular obstructive disease (Eisenmenger syndrome), others have a residual shunt and severe PAH but are not cyanotic, some patients with a small shunt may have severe PAH and are more similar to IPAH, and finally it is well known that PAH may persist and progress despite corrective cardiac surgery. Persistent pulmonary hypertension of the newborn (PPHN) is the most common cause of PH in the neonatal period, and is most often reversible (12,13). PAH associated with pulmonary veno-occlusive disease (PVOD) (14,15,16) or pulmonary capillary hemangiomatosis (PCH) (16,17,18) is also categorized in group 1. Mutations in EIF2AK4, a gene which regulates angiogenesis in response to cellular stress, have been identified both in patients with PVOD and PCH suggesting a common pathogenesis. PH associated with left heart disease (“venous hypertension”) is defined as group 2 PH. In these disorders, the main cause of PH is an elevation of left heart pressure due to impaired relaxation (diastolic dysfunction) or an anatomic lesion that leads to high left atrial pressure (such as mitral stenosis). Abnormalities of left heart inflow or outflow, such as cor triatriatum or subaortic stenosis, were recently added to the Nice classification in 2013. The initial treatment of most of these disorders generally is treatment of the anatomic obstruction as pulmonary vasodilators may lead to pulmonary edema. Of note, patients with a cavopulmonary anastomosis are not included in the Nice classification. Group 3 PH, or PH associated with lung disease or hypoxemia, is separated from group 1 as treatment of the lung disorder may result in normalization of PAP. PH associated with chronic lung diseases, such as bronchopulmonary dysplasia (BPD) (19,20,21) or congenital diaphragmatic hernia (CDH) (22,23,24,25) is increasingly recognized as an important cause of PH in the infant and child. The Nice classification includes a list of lung development abnormalities associated with PH (Table 66.5) (1). Group 4 PH, or PH associated with chronic thromboembolic disease, is a rare cause of PH in children, but is still important to recognize and treat as therapy may be curative (26). Group 5 consists of disorders with multifactorial etiology, such as SCD (27,28) or segmental PH as seen in pulmonary atresia with ventricular septal defect (VSD).
TABLE 66.1 Definitions | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
TABLE 66.2 Updated Classification of Pulmonary Hypertension | ||||||
|---|---|---|---|---|---|---|
|
TABLE 66.3 Updated Clinical Classification of Pulmonary Arterial Hypertension Associated with Congenital Heart Disease | |||||
|---|---|---|---|---|---|
|
TABLE 66.4 Cardiac Lesions Associated with Pulmonary Hypertension | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Due to the difficulties of applying the Dana Point and Nice classification to children, a working group of the Pulmonary Vascular Research Institute developed a unique pediatric classification (Table 66.6) (29). This classification recognizes the concepts of perinatal maladaptation (Fig. 66.1), maldevelopment, and
pulmonary hypoplasia as important contributors to pediatric PH disease (Fig. 66.2). Many children presenting with PH have complex heterogeneous disease consisting of prematurity, a chromosomal or genetic anomaly, a congenital cardiac defect, chronic aspiration, and sleep disordered breathing. In this classification, PH in children is defined as in adults for a biventricular circulation, but PH in the setting of single ventricle physiology is defined as a pulmonary vascular resistance index >3.0 Wood units × m2 or a transpulmonary gradient >6 mm Hg (29).
pulmonary hypoplasia as important contributors to pediatric PH disease (Fig. 66.2). Many children presenting with PH have complex heterogeneous disease consisting of prematurity, a chromosomal or genetic anomaly, a congenital cardiac defect, chronic aspiration, and sleep disordered breathing. In this classification, PH in children is defined as in adults for a biventricular circulation, but PH in the setting of single ventricle physiology is defined as a pulmonary vascular resistance index >3.0 Wood units × m2 or a transpulmonary gradient >6 mm Hg (29).
TABLE 66.5 Developmental Lung Diseases Associated with Pulmonary Hypertension | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 66.6 The broad schema of 10 basic categories of Pediatric Pulmonary Hypertensive Vascular Disease | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
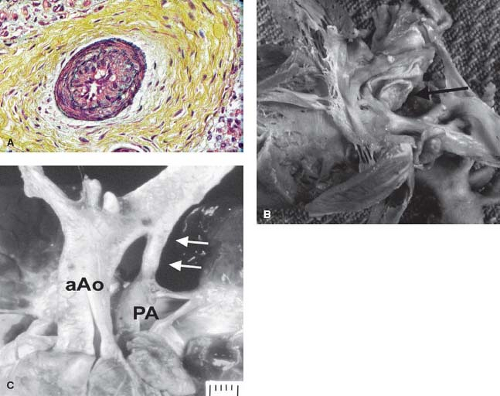 Figure 66.1 Photomicrograph of a neonate who died immediately after birth before arterial switch showing severe intimal proliferation (A) with constriction of the foramen ovale (B) (black arrow) and ductus arteriosus (C) (white arrows). aAO, ascending aorta; PA, pulmonary artery. (From Maeno YV, Kamenir SA, Sinclair B, van der Velde ME, Smallhorn JF, Hornberger LK. Prenatal features of ductus arteriosus constriction and restrictive foramen ovale in d-transposition of the great arteries. Circulation. 1999;99(9):1209–1214.) |
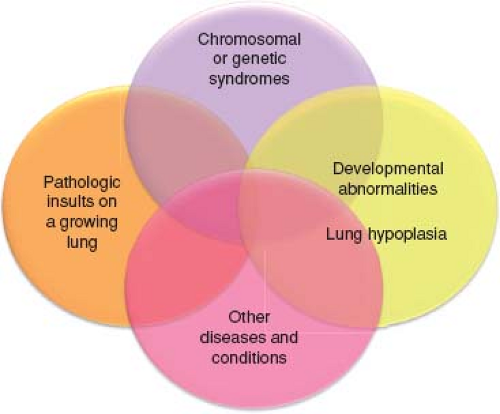 Figure 66.2 Venn diagram illustrating the heterogeneity and multifactorial elements in pediatric pulmonary hypertensive vascular disease. (From Cerro MJ, Abman S, Diaz G, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ. 2011;1(2):286–298.) |
Etiology
Over the last several years, registries have begun to identify differences in etiology between adult and pediatric PH (30,31). In children, idiopathic PAH, heritable PAH and PAH associated with CHD are the majority of causes, while PAH associated with connective tissue disease and thromboembolic PH are rare causes (10,32,33,34). The Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) registry enrolled 362 PAH patients with confirmed PAH (33). In this registry, (88%) had PAH, which was characterized as IPAH or familial PAH in 182 (57%) or associated with CHD in 115 (36%). Forty-two of the 362 patients (12%) had PH associated with respiratory disease or hypoxemia, with BPD the most frequent. Only three patients had either chronic thromboembolic PH or miscellaneous causes of PH. Chromosomal anomalies, mainly trisomy 21, were reported in 47 (13%) of patients with confirmed disease. In the national pediatric PH registry of the Netherlands, 2,845 of 3,263 pediatric patients with pulmonary hypertension had PAH (Nice group 1), including transient PAH (82%), progressive PAH (5%), or PH crisis (1%) (35). The remaining causes of PH included lung disease and/or hypoxemia (8%), PH included with left heart disease (5%), and chronic thromboembolic PH (<1%). The most common causes of transient PH were persistent PH of the neonate (PPHN) (58%) and PAH associated with CHD following repair (42%). PAH associated with repaired CHD (72%) and idiopathic PAH (23%) were common causes among the
progressive PAH cases. Down syndrome was the most frequent chromosomal disorder (12%) as was seen the result of TOPP registry. In the Spanish registry patients were classified as: PAH (n = 142, 61%), left heart disease (n = 31, 14%), respiratory disease (n = 41, 18%), thromboembolic PH (n = 2, 1%), or multifactorial (n = 10, 4.5%, mostly inherited metabolic diseases). Of the patients studied, 31% had multifactorial PH that crossed Nice WSPH groups (36).
progressive PAH cases. Down syndrome was the most frequent chromosomal disorder (12%) as was seen the result of TOPP registry. In the Spanish registry patients were classified as: PAH (n = 142, 61%), left heart disease (n = 31, 14%), respiratory disease (n = 41, 18%), thromboembolic PH (n = 2, 1%), or multifactorial (n = 10, 4.5%, mostly inherited metabolic diseases). Of the patients studied, 31% had multifactorial PH that crossed Nice WSPH groups (36).
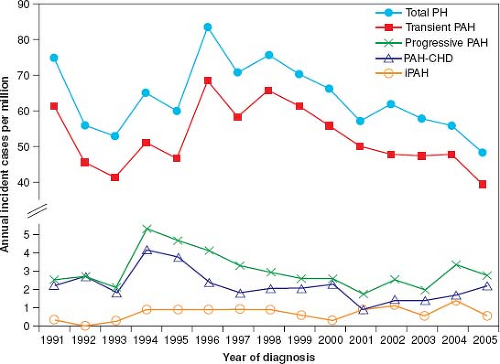 Figure 66.3 Annual incidence rates for pediatric pulmonary hypertension. PH indicates pulmonary hypertension; PAH, pulmonary arterial hypertension; PAH-CHD, PAH associated with congenital heart defects; and IPAH, idiopathic PAH. (From van Loon RL, Roofthooft MT, Hillege HL, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the period 1991 to 2005. Circulation. 2011;124(16):1755–1764.) |
Epidemiology and Survival
Until recently, the incidence and prevalence of PH in the pediatric population was not well known. In the Netherlands registry, the yearly incidence rates for PH were 63.7 cases per million children (Fig. 66.3). The annual incidence rates of idiopathic PAH and PAH associated with CHD were 0.7 and 2.2 cases per million, respectively. The point prevalence of PAH associated with CHD was 15.6 cases per million. The incidences of PPHN and transient PH associated with CHD were 30.1 and 21.9 cases per million children, respectively (35). Likewise, the incidence of idiopathic PAH in the national registries from the United Kingdom was 0.48 cases per million children per year and the prevalence was 2.1 cases per million (37). In the French registry, the estimated prevalence for chronic PAH was 3.7 cases/million (34).
Without appropriate treatments, median survival rate after diagnosis of children with IPAH might be worse compared to adults, and was 10 months for children in the NIH registry of patients with IPAH (38). In 1995, prior to the availability of targeted PAH therapies, a single-center cohort study showed the estimated median survival of children and adults with idiopathic PAH were similar (4.12 years vs. 3.12 years, respectively) (39). Currently, with targeted pulmonary vasodilators, the survival rate has continued to improve in pediatric patients with PAH. Children in the combined adult and pediatric US REVEAL registry (Registry to Evaluate Early and Long-Term PAH Disease Management) demonstrated 1-, 3-, and 5-year estimated survival rates from diagnostic catheterization of 96% ± 4%, 84% ± 5%, and 74% ± 6%, respectively (32). There was no significant difference in 5-year survival between idiopathic PAH/familial PAH (75% ± 7%) and PAH associated with CHD (71% ± 13%). Additionally, a retrospective study from the UK PH Service for Children has shown the 5-year survival in 216 children with idiopathic PAH and associated PAH (10). The survival rates of idiopathic PAH were 85.6%, 79.9%, and 71.9% at 1, 3, and 5 years, respectively, whereas associated PAH survival rates were 92.3%, 83.8%, and 56.9% at 1, 3, and 5 years, respectively. In a separate report of idiopathic PAH from the United Kingdom, survival at 1, 3, and 5 years was 89%, 84%, and 75%; while transplant-free survival was 89%, 76%, and 57% (37). Reports from the Netherlands have shown worse survival for patients with progressive PAH (not including PPHN or PAH resolving after repair of CHD) with 1, 3, and 5-year survival of 73%, 63%, and 60%, respectively (35). Children with repaired CHD may have worse outcome than other forms of PH (10).
Comparison of 275 recently diagnosed, consecutive pediatric PAH patients from three centers (Denver, New York, and Dutch) between 2000 and 2010 showed 1-, 3-, 5-, and 7-year transplantation-free survival rates of 96%, 89%, 81%, and 79%, respectively (Fig. 66.4) (40). Unadjusted survival rates differed between the center cohorts (1-, 3-, and 5-year transplantation-free survival rates: 100%, 96%, and 90% for NY; 95%, 87%, and 78% for Denver; and 84%, 71%, and 62% for Dutch, respectively; p <0.001). Based on World Health Organization (WHO) functional class and hemodynamic parameters, disease severity at diagnosis differed between the center cohorts. Adjustment for diagnosis, WHO functional class, indexed pulmonary vascular resistance, and pulmonary-to-systemic arterial pressure ratio resolved the observed survival differences.
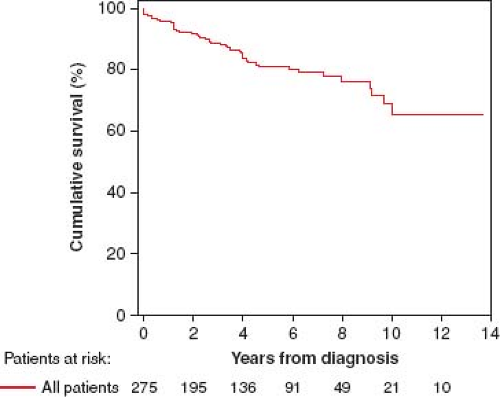 Figure 66.4 Kaplan–Meier curves showing the survival (A) for all included pediatric PAH patients: 1-, 3-, 5-, and 7-year transplantation-free survival rates were 96%, 89%, 81%, and 79%, respectively at three PH centers. PAH, pulmonary arterial hypertension. (From Zijlstra WM, Douwes JM1, Rosenzweig EB, et al. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol. 2014;63(20):2159–2169.) |
Clinical Presentation
Symptoms of PH in children are frequently misleading or subtle and the diagnosis may be unrecognized for some time. A high degree of suspicion should be the rule and PH should be suspected in any child with undue shortness of breath, tiredness, or syncope. Syncope is a common presenting symptom in children with IPAH (32,33). In patients with asthma who do not respond appropriately to medical therapy, PH should be considered in the differential diagnosis. Other clinical symptoms may include hemoptysis (41), chest pain, dizziness, arrhythmias, or in advanced disease, symptoms of right heart failure, for example, facial edema. For IPAH patients or those with CHD who have undergone complete surgical repair with elevated pulmonary vascular resistance, syncope or seizures can occur. Thus, children are sometimes misdiagnosed with a seizure disorder prior to making the diagnosis of PH. In contrast, syncope is a rare presenting symptom in patients with Eisenmenger syndrome (33).
Signs and Symptoms
The physical signs of PAH include a single and loud second heart sound without respiratory variation, right ventricular lift, and hepatomegaly with peripheral edema in the case of heart failure. In cases of severe disease, the second heart sound may be palpable. A diastolic murmur of pulmonary insufficiency, a holosystolic murmur of tricuspid regurgitation, or gallop rhythm may be audible. In infants, symptoms are less specific and may involve poor appetite, failure to thrive, lethargy, diaphoresis, tachypnea, tachycardia, and irritability (42,43,44).
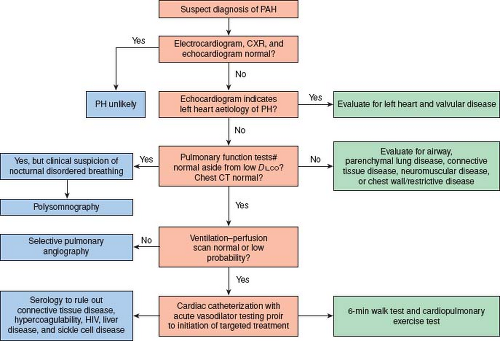 Figure 66.5 Pediatric pulmonary arterial hypertension diagnostic workup. (Adapted from Rosenzweig EB, Feinstein JA, Humpl T, Ivy DD. Pulmonary arterial hypertension in children: diagnostic work-up and challenges. Prog Pediatr Cardiol. 2009;27:4–11.) |
Evaluation
Due to the many diseases associated with PH, a methodical and comprehensive evaluation is important (Fig. 66.5) (45). The most successful strategy in treatment of PH is correction of an underlying abnormality, rather than addition of vasodilator therapy. Special situations may predispose to the development of PAH. Children living at altitude and presenting with high-altitude pulmonary edema (HAPE) should be screened for PH (46). In addition, children with biliary atresia, cavernous transformation of the portal vein, primary sclerosing cholangitis, or cryptogenic cirrhosis, may have portopulmonary hypertension (PPHTN) with an associated high mortality (47,48,49). The development of pulmonary edema in response to pulmonary vasodilators suggests diseases such as PCH, PVOD, or in infants, nonketotic hyperglycinemia (50). Deficiencies of vitamin C (51) and cobalamin C (52,53,54) have also been associated with PH.
Chest Radiography
The enlargement of the central pulmonary artery and/or right ventricle on chest radiography suggests the presence of PAH. Prominence of the main pulmonary arteries is apparent in most patients with IPAH or portal hypertension. The chest radiograph may show variable peripheral lung fields, depending on the amount of pulmonary blood flow. As pulmonary blood flow decreases as a result of increasing pulmonary vascular resistance, the lung fields become progressively oligemic. Chest radiography findings may be useful to uncover secondary causes of PH. Pulmonary venous congestion may suggest PVOD or PCH; hyperinflation or kyphosis are signs of restrictive lung disease; asymmetry of the enlarged central pulmonary arteries may warrant investigation of chronic thromboembolic disease or PPHTN (44,55,56,57). Asymmetric lung volumes may suggest either pulmonary arterial or pulmonary venous abnormalities. A unilateral small lung may be seen in isolated pulmonary artery of ductal origin (also known as “unilateral absence of a pulmonary artery”) (58), Scimitar syndrome (59,60) or unilateral congenital absence of pulmonary veins.
Electrocardiography
Electrocardiography (ECG) is often the first test to suggest PH by showing right ventricular hypertrophy and right atrial enlargement. Evidence of right ventricular hypertrophy on ECG is present in most but not all children with PH. ECG findings include a qR complex in lead V1 or V3R regardless of voltage. An upright T wave in V1 is indicative of right ventricular hypertrophy from 7 days to 7 years. However, some studies have suggested that the specificity (69%) and positive predictive value (67%) of EKG are low in children with an echocardiographic diagnosis of PH (61). However, the sensitivity is likely higher, especially in combination with a complete physical examination.
Echocardiography
Echocardiography is the most useful noninvasive screening tool to evaluate patients with a clinical suspicion of PAH (62). The echocardiogram documents cardiac anatomy, right ventricular size and function, left ventricular systolic and diastolic function, morphology and function of valves, and the presence of pericardial effusion or a patent foramen ovale. Doppler ultrasound can be used noninvasively to estimate the pulmonary artery systolic pressure (PASP). PASP can be determined by measuring the peak systolic
pressure gradient from the right ventricle to the right atrium and calculated using the Bernoulli equation (4v2, where v is the maximum velocity of the TR jet measured by continuous wave Doppler and added to the estimated right atrial pressure) (Fig. 66.6). One must be aware that using the tricuspid regurgitant jet velocity as an estimate of PASP can over or underestimate the PAP and thus other features of the echocardiogram are essential to support the diagnosis of PH (63,64). The presence of right ventricular outflow tract obstruction also invalidates the TR jet as an accurate estimate of PASP. A qualitative assessment of RV function is also important. This is often challenging due to the geometry of the RV which is more complex to analyze than the left ventricle. Several measures are available to attempt to quantify the degree of RV dysfunction including the Tei index (myocardial performance index), RV ejection fraction, RV fractional area change, and the tricuspid annular plane systolic excursion (TAPSE) (65,66,67,68,69,70,71,72). Normal
values for TAPSE in children have recently been published and should serve as a reference for children with PH (Fig. 66.7) (68). The ratio of right ventricle to left ventricle size at end systole is a strong predictor of outcome (Fig. 66.8) (73). An increasing RV/LV systolic ratio is associated with an increasing hazard for a clinical event (hazard ratio, 2.49; 95% confidence interval, 1.92 to 3.24). Pulmonic valve insufficiency is frequently seen, and characteristics of the pulmonic regurgitant flow velocity or changes in the systolic flow velocity profile across the pulmonic valve also can be used to estimate noninvasively the pulmonary artery diastolic pressure and the mean PAP (see Fig. 66.6) (74). The presence of a pericardial effusion is rare in children, but when present, suggests a poor prognosis (69,75). As PH progresses and RV function worsens the systolic portion of the cardiac cycle lengthens leading to an increase in the systolic: diastolic (S:D) ratio. The S:D ratio is higher in PH patients than in controls (1.38 ± 0.61 vs. 0.72 ± 0.16, p <0.001), and is associated with worse echocardiographic RV fractional area change, worse catheterization hemodynamics, shorter 6-minute walk distance (6MWD), and worse clinical outcomes independent of pulmonary resistance or pressures (Fig. 66.9) (72,76,77). Tissue Doppler imaging (TDI) directly measures myocardial velocities and has been shown to be an accurate measure of RV and LV systolic and diastolic function. In recent pediatric studies, right ventricular TDI velocity was lower in children with PAH compared to healthy controls (78,79). Moreover, tricuspid diastolic velocity (E′) had significant inverse correlations with right ventricular end-diastolic pressure and mean pulmonary arterial pressure, and cumulative event-free survival rate was significantly lower when tricuspid E′ velocity was ≤8 cm/s (log-rank test, p <0.001, Fig. 66.10) (79). Through ventricular interdependence, PH induces left ventricular dysfunction; reduced LV global longitudinal strain is reduced predominantly because of reduced basal and mid septal strain (80). The right ventricle contracts primarily in a longitudinal fashion, thus RV longitudinal strain measurement may play an important role in evaluation of RV function. RV longitudinal strain is a powerful tool to predict clinical outcome in adults with PH (81). Finally, real-time three-dimensional echocardiography correlates well with cardiac MRI in children with CHD (82) and is being evaluated in children with PH.
pressure gradient from the right ventricle to the right atrium and calculated using the Bernoulli equation (4v2, where v is the maximum velocity of the TR jet measured by continuous wave Doppler and added to the estimated right atrial pressure) (Fig. 66.6). One must be aware that using the tricuspid regurgitant jet velocity as an estimate of PASP can over or underestimate the PAP and thus other features of the echocardiogram are essential to support the diagnosis of PH (63,64). The presence of right ventricular outflow tract obstruction also invalidates the TR jet as an accurate estimate of PASP. A qualitative assessment of RV function is also important. This is often challenging due to the geometry of the RV which is more complex to analyze than the left ventricle. Several measures are available to attempt to quantify the degree of RV dysfunction including the Tei index (myocardial performance index), RV ejection fraction, RV fractional area change, and the tricuspid annular plane systolic excursion (TAPSE) (65,66,67,68,69,70,71,72). Normal
values for TAPSE in children have recently been published and should serve as a reference for children with PH (Fig. 66.7) (68). The ratio of right ventricle to left ventricle size at end systole is a strong predictor of outcome (Fig. 66.8) (73). An increasing RV/LV systolic ratio is associated with an increasing hazard for a clinical event (hazard ratio, 2.49; 95% confidence interval, 1.92 to 3.24). Pulmonic valve insufficiency is frequently seen, and characteristics of the pulmonic regurgitant flow velocity or changes in the systolic flow velocity profile across the pulmonic valve also can be used to estimate noninvasively the pulmonary artery diastolic pressure and the mean PAP (see Fig. 66.6) (74). The presence of a pericardial effusion is rare in children, but when present, suggests a poor prognosis (69,75). As PH progresses and RV function worsens the systolic portion of the cardiac cycle lengthens leading to an increase in the systolic: diastolic (S:D) ratio. The S:D ratio is higher in PH patients than in controls (1.38 ± 0.61 vs. 0.72 ± 0.16, p <0.001), and is associated with worse echocardiographic RV fractional area change, worse catheterization hemodynamics, shorter 6-minute walk distance (6MWD), and worse clinical outcomes independent of pulmonary resistance or pressures (Fig. 66.9) (72,76,77). Tissue Doppler imaging (TDI) directly measures myocardial velocities and has been shown to be an accurate measure of RV and LV systolic and diastolic function. In recent pediatric studies, right ventricular TDI velocity was lower in children with PAH compared to healthy controls (78,79). Moreover, tricuspid diastolic velocity (E′) had significant inverse correlations with right ventricular end-diastolic pressure and mean pulmonary arterial pressure, and cumulative event-free survival rate was significantly lower when tricuspid E′ velocity was ≤8 cm/s (log-rank test, p <0.001, Fig. 66.10) (79). Through ventricular interdependence, PH induces left ventricular dysfunction; reduced LV global longitudinal strain is reduced predominantly because of reduced basal and mid septal strain (80). The right ventricle contracts primarily in a longitudinal fashion, thus RV longitudinal strain measurement may play an important role in evaluation of RV function. RV longitudinal strain is a powerful tool to predict clinical outcome in adults with PH (81). Finally, real-time three-dimensional echocardiography correlates well with cardiac MRI in children with CHD (82) and is being evaluated in children with PH.
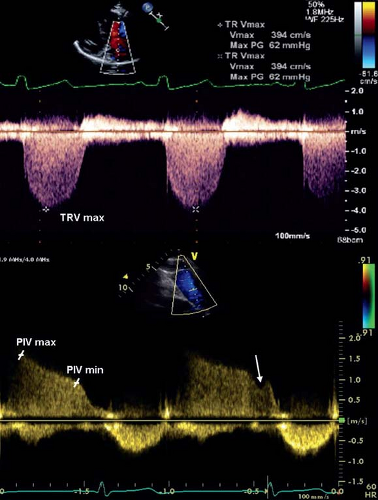 Figure 66.6 Echocardiographic evaluation of pulmonary hypertension. Spectral Doppler images of tricuspid regurgitation (TR), demonstrating an optimal Doppler envelope to obtain the maximal TR velocity (top image), and of pulmonary insufficiency (bottom image). The left marker represents the peak early-diastolic velocity and the right marker represents the end-diastolic velocity of the pulmonary insufficiency Doppler curve. The arrow represents the end-diastolic “shoulder” of the pulmonary insufficiency jet. TRV max, tricuspid regurgitation peak velocity; PIV max, pulmonary insufficiency peak velocity; PIV min, pulmonary sufficiency end-diastolic velocity. (From Tissot C, Ivy DD. In: Beghetti M, ed. Pediatric Pulmonary Hypertension. Munich: Elsevier Urban & Fischer; 2011.) |
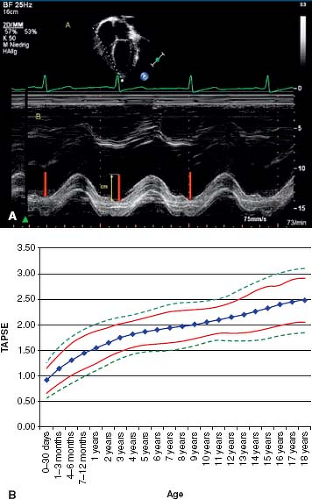 Figure 66.7 Measurement (A) and normal values for tricuspid annular plane systolic excursion (B) (TAPSE). (From Koestenberger M, Ravekes W, Everett AD, et al. Right ventricular function in infants, children and adolescents: reference values of the tricuspid annular plane systolic excursion (TAPSE) in 640 healthy patients and calculation of z score values. J Am Soc Echocardiogr. 2009;22(6):715–719.) |
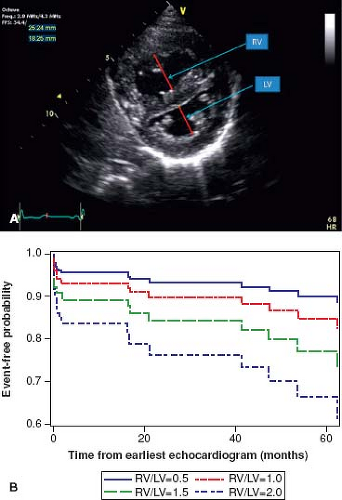 Figure 66.8 Parasternal short-axis view of the right and left ventricles at the level of the papillary muscles (A). The RV/LV ratio is derived from RV diameter and LV diameter at end-systole. RV/LV end-systole ratio is predictive of outcome. Estimated survival curves for four possible RV/LV ratios estimated from the Cox varying coefficients regression corresponding to a hazard ratio of 2.49 for RV/LV ratio (B). (From Jone PN, Hinzman J, Wagner BD, Ivy DD, Younoszai A. Right ventricular to left ventricular diameter ratio at end-systole in evaluating outcomes in children with pulmonary hypertension. J Am Soc Echocardiogr. 2014;27:172–178.) |
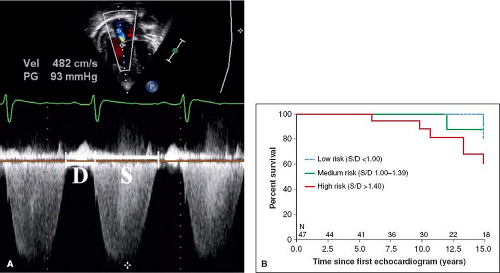 Figure 66.9 The systolic (S) to diastolic (D) time ratio from tricuspid regurgitation velocity (A) can be measured as a measure of right ventricular function. An increase in the S/D ratio predicts worse outcome in children with PH (B). (From Alkon J, Humpl T, Manlhiot C, McCrindle BW, Reyes JT, Friedberg MK. Usefulness of the right ventricular systolic to diastolic duration ratio to predict functional capacity and survival in children with pulmonary arterial hypertension. Am J Cardiol. 2010;106:430–436.) |
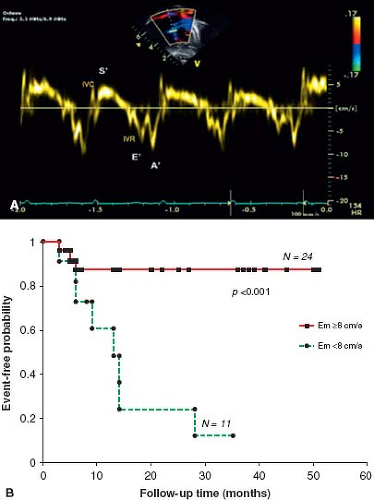 Figure 66.10 Tissue Doppler imaging of the right ventricle at the lateral annulus of the tricuspid valve measures the myocardial systolic wave (S’) which measures the systolic longitudinal function of the RV and two diastolic waves: early diastolic (E’) and late diastolic (A’, which denote the diastolic function of the ventricles(A). Low E’ velocity less than 8 cm/s is predictive of poor outcome in pediatric IPAH (B). (From Takatsuki S, Nakayama T, Jone PN, et al. Tissue Doppler imaging predicts adverse outcome in children with idiopathic pulmonary arterial hypertension. J Pediatr. 2012;161(6):1126–1131.) |
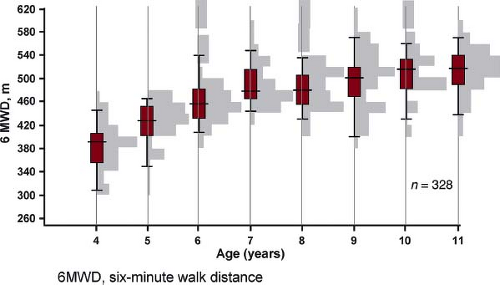 Figure 66.11 The distance walked in 6 minutes by different age groups in normal children. Data are shown as histograms and box- and whiskers-plots illustrating the median, 25th and 75th percentile (gray box) and 5th and 95th percentile. (From Lammers AE, Hislop AA, Flynn Y, Haworth SG. The 6-minute walk test: normal values for children of 4–11 years of age. Arch Dis Child. 2008;93:464–468.) |
Cardiac Mri
Cardiac MRI provides exact measurements of heart function and blood flow and is used with increasing frequency to evaluate patients with PAH (83,84,85,86,87,88,89,90). Cardiac MRI provides valuable information without radiation on right ventricular volumes, mass, left ventricular septal bowing, cardiac index, and the presence of delayed contrast enhancement. Most young children require anesthesia for this procedure which is an important consideration. A recent study of 100 children with PH has shown that of the cardiac MRI parameters measured, right ventricular ejection fraction and left ventricular stroke volume index were most strongly predictive of survival on univariate analysis (2.6- and 2.5-fold increase in mortality for every 1-SD decrease, respectively) (91). Septal curvature measured by cardiac MRI correlates strongly with mean PAP at baseline and during vasodilator testing (92).
Exercise Capacity
Cardiopulmonary exercise testing (CPET) using cycle ergometry or 6-minute walk testing (6MWT) has been shown to correlate with disease severity and prognosis, and is helpful in assessing responses to clinical treatments. Recently published data showed that children can safely undergo cardiopulmonary testing and the peak oxygen consumption is strongly correlated to disease severity (93,94,95). A recent study of maximal CPET using cycle ergometry in children with PH concluded that the study could be performed safely in pediatric patients with PH, with the exception of patients with severe limitation who were excluded from exercise testing. ST segment depression was graded as mild (19%) or moderate (1.5%). There were no significant adverse events, such as syncope, chest pain, or dizziness, and the study was stopped for fatigue in 53% of patients, leg fatigue in 23%, dyspnea in 21%, and miscellaneous reasons in 3% (95). Additional exercise parameters, such as the ventilatory efficiency ratio (VE/VCO2) correlates well with invasive parameters and was predictive of outcome (ratio 51.1 poor outcome vs. 37.9 good outcome, p <0.001) (96).
6MWT is a submaximal test shown to have a strong independent association with mortality among adults with IPAH (97,98,99,100,101,102); however, children often have less right heart failure for a given elevation of PAP leading to a further distance walked. A recent study compared CPET with 6MWD. Peak VO2 and oxygen consumption at anaerobic threshold showed correlation with 6MWD (r = 0.49; p = 0.001 and r = 0.40, p = 0.01, respectively), while an inverse correlation was found between measures of ventilatory efficiency (VE/VCO2) at anaerobic threshold and 6MWT distance (r = −0.43; p = 0.005). The 6MWT reflected maximal exercise capacity in children with a 6MWT distance below 300 m, but correlation was poor above 300 m (101). Normal values for 6MWD for children have recently been published (Fig. 66.11) (98,99,100,102). However baseline 6MWD is not a predictor of survival, neither when expressed as an absolute distance in meters nor when adjusted to reference values expressed as z-score or as percentage of predicted value (37,103).
Cardiac Catheterization and Biomechanical Evaluation
Cardiac catheterization is important in the evaluation of PH as it accurately measures PAP and resistance, evaluates the presence and severity of CHD, provides prognostic information on outcome, and provides information on operability in CHD and on initial vasodilator therapy in PAH. The definition of a “responder” at catheterization is different for patients being evaluated for operability and those being evaluated for acute vasodilator challenge to determine initial vasodilator therapy. For PH therapy, it is uncertain if the same definition of vasoreactivity should be applied for adults and children. An anesthesiologist experienced in the management of PH should be present during the cardiac catheterization, as sedation may pose a significant risk to the patient with PAH (104,105,106,107). A skilled specialist with experience doing cardiac catheterizations on children with PAH should perform the procedure. Patients with suprasystemic PH pose particular risk (106,108). In the TOPP registry complications associated with heart catheterization (HC) were analyzed in a total of 908
HCs; 554 were at diagnosis and 354 in follow-up. Complications were reported in 5.9% with five deaths considered related to HC, suggesting a higher rate of HC complications compared to adult studies (107). During the catheterization, baseline measures including right atrial pressure, pulmonary arterial pressure, systemic arterial pressure, mixed venous and systemic arterial saturation, cardiac index, pulmonary capillary wedge pressure, PVR, SVR, and PVR/SVR ratio should be obtained in addition to evaluation of shunts when there is a suspicion of a systemic to pulmonary communication (109). Measurements must be obtained when the patient is closest to their usual hemodynamic and metabolic state and with a normal pH. Particular care is necessary at the time of catheterization to exclude additional intracardiac as well as extracardiac defects and to measure left ventricular filling pressure accurately to rule out postcapillary Pulmonary hypertension. Acute vasodilator testing is essential at the baseline diagnostic catheterization to assist in determining the optimal therapeutic regimen, and is described in a subsequent section. In children with the most severe pulmonary vascular disease, catheterization may be delayed until the child is stabilized.
HCs; 554 were at diagnosis and 354 in follow-up. Complications were reported in 5.9% with five deaths considered related to HC, suggesting a higher rate of HC complications compared to adult studies (107). During the catheterization, baseline measures including right atrial pressure, pulmonary arterial pressure, systemic arterial pressure, mixed venous and systemic arterial saturation, cardiac index, pulmonary capillary wedge pressure, PVR, SVR, and PVR/SVR ratio should be obtained in addition to evaluation of shunts when there is a suspicion of a systemic to pulmonary communication (109). Measurements must be obtained when the patient is closest to their usual hemodynamic and metabolic state and with a normal pH. Particular care is necessary at the time of catheterization to exclude additional intracardiac as well as extracardiac defects and to measure left ventricular filling pressure accurately to rule out postcapillary Pulmonary hypertension. Acute vasodilator testing is essential at the baseline diagnostic catheterization to assist in determining the optimal therapeutic regimen, and is described in a subsequent section. In children with the most severe pulmonary vascular disease, catheterization may be delayed until the child is stabilized.
Recent interest has grown in the area of measurement of total right ventricular afterload by measurement of input vascular impedance (110,111,112,113). Impedance incorporates both resistive (static) and capacitive (pulsatile) components of the vascular bed (113,114,115,116,117). Currently, measurement of impedance requires invasive measurements in addition to measurement of Doppler flow. A recent manuscript in adults with PH showed that a measure of capacitance (stroke volume/pulse pressure) was a better predictor of survival than measurement of pulmonary vascular resistance (118,119). In children, pulsatile components of right ventricular afterload, represented by pulmonary arterial capacitance and pulmonary stroke volume index, provide important prognostic information to conventional static hemodynamic parameters (120,121). In children pulmonary vascular input impedance has recently been shown to be feasible and predict clinical outcomes better than PVR in children with PAH (122).
In the case of the right ventricle, ventricular vascular coupling (VVC) refers to the RV functional adaptation to afterload (123). Measures of VVC correlate with outcome in adult PH (124). RV stroke work (RVSW), the product of mean PAP and stroke volume, integrates contractility, afterload and VVC. RVSW can be estimated in children with PAH by echocardiography or catheterization, and is significantly associated with abnormal WHO functional class, the need for atrial septostomy, as well as mortality (125).
Laboratory Assessment
A comprehensive serologic evaluation looking for other systemic conditions that might lead to PAH and warrant additional treatment is performed (Table 66.7). Approximately 5% to 20% of children with idiopathic disease may have an elevation of antinuclear antibodies as well as evidence of hypothyroidism or hyperthyroidism suggesting an autoimmune association (126,127,128,129). Furthermore, children should be screened for evidence of a hypercoagulable state as some may have an underlying coagulopathy such as antiphospholipid antibody syndrome or a lupus anticoagulant. The evaluation should include a liver ultrasound in cases suspect of having PPHTN. A chest CT scan is important in the evaluation of the child with suspected IPAH to be certain that there is no underlying interstitial lung disease. CT scan of the chest may also be very helpful in evaluation for PCH or veno-occlusive disease. HIV and sickle cell testing should be routinely performed in those patients at risk. If there is a family history of PH, early/sudden death, or severe disease in a young child genetic testing/counseling may be beneficial.
TABLE 66.7 Laboratory Analyses for PAH Workup | |||||||
|---|---|---|---|---|---|---|---|
|
Ventilation–Perfusion Scintigraphy
Ventilation–perfusion lung scanning is important in patients in whom IPAH is suspected to exclude chronic thromboembolic disease as a cause of the elevated PAP. Although thromboembolic disease is rare in children, it is one of the few diseases that can be cured with appropriate therapy including pulmonary thromboendarterectomy (26). A normal or low-probability lung scan essentially rules out thromboembolic disease as the cause of the PH. Patients with IPAH will frequently have a patchy appearance to the perfusion scan.
Pulmonary Function Testing/Nocturnal Oximetry
Evaluation for obstructive or restrictive lung disease should be performed as part of the evaluation of children with PH. Abnormalities of pulmonary function may be present in PAH patients particularly in the more advanced stages of the disease. A mild restrictive lung defect is seen in 20% to 50% of patients with IPAH. The presence of moderate or severe restrictive or obstructive physiologic defects should suggest another diagnosis. Mild reduction in the diffusion capacity is seen in most patients with IPAH. Severe hypoxemia is not usually present unless there is either intracardiac shunting via a patent foramen ovale or an open congenital heart defect in patients with Eisenmenger syndrome or severely depressed cardiac output with resultant mixed venous hypoxemia in both conditions. Nocturnal oximetry should be performed as
a screening test for obstructive sleep apnea (OSA) or sleep disordered breathing if there is a suspicion of OSA, for example, in a child with enlarged tonsils or adenoids and history of snoring. Sleep disordered breathing may cause mild PH which may resolve with night time CPAP.
a screening test for obstructive sleep apnea (OSA) or sleep disordered breathing if there is a suspicion of OSA, for example, in a child with enlarged tonsils or adenoids and history of snoring. Sleep disordered breathing may cause mild PH which may resolve with night time CPAP.
Evaluation of Reflux and Aspiration
In the young child, reflux and aspiration may worsen PH. Many children with genetic syndromes, and in particular Down syndrome, routinely have aspiration. Silent aspiration is of particular concern, as these children do not always cough during aspiration events. Modified barium swallow study and measurement of reflux with a pH probe are important studies in the evaluation of these patients. In general, treatment of reflux and aspiration should precede treatment with pulmonary vasodilators.
Biomarkers and Treatment Goals
Interest in biomarkers has grown in the last several years. In adults, b-type natriuretic peptide (BNP) is a useful tool to assess mortality risk, progression of the disease, and response to therapy (130). Recent studies in children have begun to identify usefulness of BNP or N-terminal pro-BNP (NT-proBNP) (131,132,133). BNP correlates positively with functional status in children with PAH and values above 130 pg/mL are associated with increased risk of death or need for transplantation (132). Furthermore, change in BNP measurements over time correlates with the change in the hemodynamic and echocardiographic parameters of children with PAH; with a BNP value >180 pg/mL predicting a decreased survival rate (Fig. 66.12). The change in BNP level in a specific patient over time was shown to be more helpful in determining risk or hemodynamic response to therapy than the average BNP value in a pediatric PAH population (131). NT-proBNP, uric acid, or norepinephrine levels have also been shown to correlate with outcome in children with PAH. A meta-analysis of studies in children with PAH found that in addition to NTproBNP, WHO functional class, mean right atrial pressure, cardiac index, indexed pulmonary vascular resistance, and acute vasodilator response were significant prognostic indicators (133).
Several biomarkers may be used as treatment goals; however, to be useful treatment-induced improvements in these variables should be associated with improved survival. In the Netherlands national registry, only the variables WHO-FC (World Health Organization-Functional Class), NT-proBNP, and TAPSE were identified as follow-up predictors in which treatment-induced changes were associated with survival. Patients in whom these variables improved after treatment showed better survival (p <0.002) (134).
Innovative approaches have begun to evaluate for novel biomarkers (135) and have implicated a role for inflammation in pediatric PH. Using a nonbiased gel-based proteomic analysis, differences in plasma proteins known to modulate inflammation, were found between children with a good or poor outcome to chronic therapy (136). Before and after therapy, serum amyloid A-4 (an acute phase protein released in response to inflammatory stimuli) was fourfold higher in those with poor outcome (death, initiation of intravenous prostacyclin) compared to those with good outcome (survival, discontinuation of intravenous prostacyclin) (136). Interleukin-6, a proinflammatory cytokine, is associated with the occurrence of an adverse event in pediatric PH (137). Inflammatory cells, such as fibrocytes (characterized by expression of CD45-leukocytes and collagen) are recruited to sites of injury in persistent inflammatory remodeling and proangiogenic signaling. Similarly, myeloid-derived suppressor cells (MDSCs) are increased in inflammatory disease and orchestrate immune cell responses. Recently published studies have shown that circulating fibrocytes and MDSCs were increased in 26 children with PAH compared to non-PAH controls (138,139). High levels of tissue inhibitors of metalloproteinases-1 (TIMP-1), which is overexpressed by proinflammatory cytokines, and low levels of apolipoprotein-A1 (140), that reduces levels of oxidized lipids and improve vascular disease are strongly associated with outcome in pediatric PH (Fig. 66.13) (141).
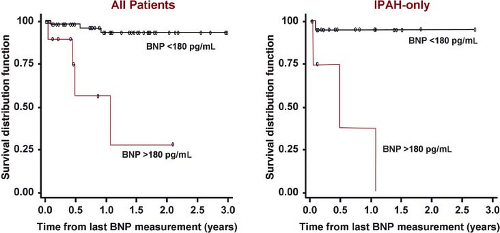 Figure 66.12 Kaplan–Meier survival curves for children with IPAH and PAH associated with CHD. Survival curves are shown for all patients (left) and for the subgroup of IPAH patients (right) categorized with either brain natriuretic peptide (BNP) >180 pg/mL or <180 pg/mL. (From Bernus A, Wagner BD, Accurso F, Doran A, Kaess H, Ivy DD. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest. 2009;135:745–751.) |
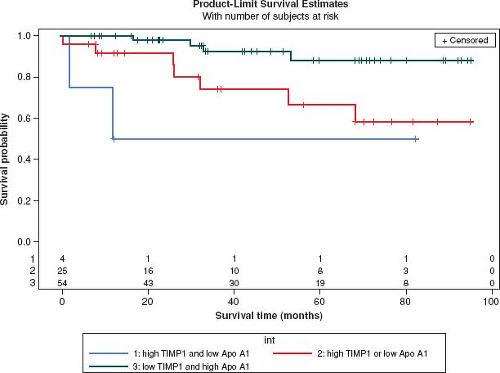 Figure 66.13 High levels of tissue inhibitors of metalloproteinases-1 (TIMP-1), which is overexpressed by proinflammatory cytokines, and low levels of apolipoprotein-A1, which reduces levels of oxidized lipids and improves vascular disease are strongly associated with outcome in pediatric PH. Patients with high TIMP-1 and low apolipoprotein-A1 values (red) had lower survival (log-rank test p value, 0.01). The number of subjects at-risk is displayed along the x-axis. (From Wagner BD, Takatsuki S, Accurso FJ, Ivy DD. Evaluation of circulating proteins and hemodynamics towards predicting mortality in children with pulmonary arterial hypertension. PLoS One. 8(11):e80235.) |
Lung Biopsy and Wedge Angiography
Lung biopsy is not used as part of the routine workup of PH given the relatively high risk of the procedure in children with PH. Performance of a lung biopsy has played a major role in understanding PH associated with CHD as is described in Chapter 65, and may help guide surgical management in select cases (142,143,144,145,146). In a series of children with CHD and PH assessed 1 year after repair, Rabinovitch et al. compared pulmonary hemodynamics and morphometric changes. Hemodynamics normalized in all children repaired before 9 months of age, regardless of severity of the preoperative Heath–Edwards, morphometric, or hemodynamic changes. In contrast, in children with severe preoperative changes at lung biopsy (grade C morphometric, Heath–Edwards III), PAP and PVR remained significantly elevated if they were repaired after 2 years of age (146). The Heath–Edwards changes are heterogeneous making absolute determination of being inoperable difficult (147).
Lung biopsy is indicated if there is a suspicion of another diagnosis which would be treated differently such as PVOD or PCH. Likewise, an increasing number of patients are being diagnosed with interstitial lung disease associated with PH. Examples include surfactant protein deficiency, such as ATP-binding cassette transporter A3 (ABCA3) deficiency, or capillaritis (148,149,150,151). These patients often do not respond to treatments currently available for PAH and thus need a definitive diagnosis for lung transplant listing.
Important Causes of PH in Children
Idiopathic Pulmonary Arterial Hypertension
PAH was first described by Dresdale in 1951 as a sporadic disease known as primary PH. Subsequently, in 1954, Dresdale reported the familial transmission of PPH. IPAH, previously called primary PH, is characterized by progressive and sustained elevations of PAP without a defined etiology. IPAH is a rare disease which occurs most frequently in young adult females (154). While generally developing in the adult population, pediatric IPAH is well reported, and carried a dismal prognosis in the NIH cohort, with a median survival of only 10 months in patients less than 16 years old (38). Evaluation for IPAH in the pediatric age group is similar to that outlined for adults and is a diagnosis of exclusion, but increased scrutiny for the possibility of congenital cardiac disease or developmental lung diseases is appropriate, and acute pulmonary vasoreactivity may be more common in children (37,155,156,157). Certain comorbidities, such as thyroid disease, an elevated ANA and lower airways obstruction are seen in children with IPAH (127,129,158).
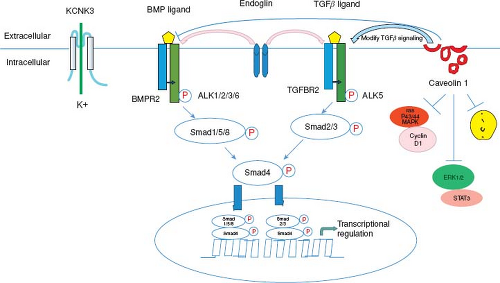 Figure 66.14 Molecular pathogenesis of hereditary PAH. BMP binds to BMPR2. Upon ligand binding, the type II receptor phosphorylates a type I receptor, including ALK1. This leads to phosphorylation of SMAD1/5/8 and phosphorylation of SMAD4 with translocation of the phosphorylated Smads to the nucleus to modulate the expression of target genes. Upon TGFβ ligand binding, the TGFβ type II receptor phosphorylates a type I receptor, ALK5. This leads to phosphorylation of SMAD2/3 which phosphorylate SMAD4 and translocate to the nucleus. Endoglin is an accessory membrane glycoprotein that interacts with signaling receptor complexes for the BMP and TGFβ superfamily. Caveolin-1 functions as a tonic inhibitor of eNOS to facilitate NO-mediated relaxation. Caveolin-1 modifies TGFβ signaling at the plasma membrane which may provide a mechanistic link between CAV1 and BMPR2 mutations. KCNK3 is a potassium channel protein in pulmonary artery smooth muscle cells. Activation of K+ channel causes vasodilatation. (Modified from Ma L, Chung WK. The genetic basis of pulmonary arterial hypertension. Hum Genet. 2014;133:471–479.) |
Heritable–Familial Pulmonary Artery Hypertension
Understanding the genetic causes of PAH has rapidly advanced in the last 5 years with seven genes presently implicated in the disease (Fig. 66.14) (159,160,161). Between 6% and 12% of cases of IPAH may be familial in origin with an autosomal dominant pattern of inheritance (160,161). BMPR2 mutations are the most common mutation and have been identified in children and adults with IPAH and familial PAH (162,163,164,165,166). This genetic mutation in the TGF-beta receptor family has been found in patients with familial PAH (50% to 80%) (162,164) and sporadic PAH (15% to 26%) (165). Currently about 300 mutations in the BMPR2 gene have been discovered (167). The pattern of inheritance in children with BMPR2 mutations is the same as in adults with an autosomal dominant pattern with reduced penetrance. Reduced penetrance implies that generations of mutation-carrying persons may not express the disease. Thus, only about 20% of individuals with a known BMPR2 mutation will develop clinical PAH. Advanced gene-sequencing methods have facilitated the discovery of additional genes with mutations among those with and those without familial forms of PAH (SMAD9, CAV1, KCNK3, EIF2AK4, TBX4) (159,160,161,168). The reduced penetrance, variable expressivity, and female predominance of PAH suggest that genetic, genomic, and other factors modify disease expression (160,161).
The bone morphogenetic proteins (BMPs) are members of the TGF-beta superfamily of receptors. BMPs play an important role in the development of the lungs, bone, and cartilage. BMP binds to the BMPR2 receptor leading to heterodimerization with the BMPR1 receptor. Activation of the receptor leads to phosphorylation of a series of cytoplasmic mediators, such as the SMAD proteins. In the BMP cascade, SMAD proteins 1/5/8 complex with SMAD4 for translocation to the nucleus leading to target gene expression and inhibit cell growth and induction of apoptosis (see Fig. 66.14) (162,169,170,171,172,173,174,175,176). A similar cascade is present for TGF-beta signaling. It has been proposed that a second hit leads to a further decrease in BMP signaling thus creating an imbalance/increase in pro-proliferative TGF-beta signaling.
BMPR2 mutations have been evaluated in several pediatric series with inconsistent results. Grunig found no BMPR2 mutations or deletions in 13 children with IPAH (163). However, in a study by Harrison et al., 22% of children with IPAH or PH associated with CHD had activin-like kinase type-1 (ALK-1) or BMPR2 mutations (177). BMPR2 mutations were found in only 6% of a mixed cohort of adults and children with PAH and CHD (178). More recently, a study by Rosenzweig et al. evaluated whether children and adults with PAH had a positive response to acute vasodilator testing. In this study, the authors found that BMPR2 mutation positive children appeared less likely to respond to acute vasodilator testing than mutation negative children (179). Furthermore, these children had lower mixed venous saturation and cardiac index than BMPR2 mutation negative patients. A limitation of this study was that there were very few children who were positive for the BMPR2 mutation.
These findings are similar to those by Elliott et al. who reported that IPAH and FPAH adult patients with BMPR2 mutations are less likely to respond to acute vasodilator testing than BMPR2 mutation negative patients (180). Recently Sztrymf reported that patients with a BMPR2 mutation presented at an earlier age, developed more severe disease and died earlier (181). Mutations in other members of the TGF-beta family, such as ALK-1 or endoglin have also been described as leading to PH in children (167,172,182,183).
These findings are similar to those by Elliott et al. who reported that IPAH and FPAH adult patients with BMPR2 mutations are less likely to respond to acute vasodilator testing than BMPR2 mutation negative patients (180). Recently Sztrymf reported that patients with a BMPR2 mutation presented at an earlier age, developed more severe disease and died earlier (181). Mutations in other members of the TGF-beta family, such as ALK-1 or endoglin have also been described as leading to PH in children (167,172,182,183).
The variable clinical expression with reduced penetrance makes genetic counseling difficult. As there is reduced penetrance, only 20% of carriers will develop the disease. A negative BMPR2 testing result may be reassuring for a family member with a known BMPR2 mutation. If negative, a family member’s risk falls from 1 in 10 to that of the general population, which is about 1 in 1,000,000. It is mandatory that genetic testing be performed in conjunction with experienced genetic counselors before and after the test results (162).
Other genetic loci may also play important roles. Studies have suggested an important role of the serotonin transporter gene in some adults with PAH (184,185), and a study in children found that homozygosity for the long variant of the serotonin transporter gene was highly associated with idiopathic PH in children (186).
Connective Tissue Disease
PAH complicates the course of some patients with CTD. The recent WHO classification recognizes PAH associated with CTD in group 1 PAH and CTD as one of the most common forms of PAH in adults. It is estimated that almost 30% of adult cases of PAH are associated with CTD, including 10% to 15% with systemic sclerosis (187). Among these patients, PAH is often multifactorial, including pulmonary fibrosis–related CTD and thrombosis associated with vasculitis and/or thrombophilia. PAH associated with CTD is also a major risk factor for death in these patients. Recently, various pulmonary vasodilator therapies have improved the prognosis for patients with PAH associated with CTD. Previous studies have been shown that epoprostenol increased exercise capacity compared with conventional therapy in patients with PAH associated with systemic sclerosis (188) and improves hemodynamics and exercise capacity in patients with systemic lupus erythematosus (189). Pulmonary vasodilators have been used in these patients with some success. Management of the underlying CTD is crucial for successful management of the child with PAH and CTD (190).
Congenital Heart Disease
A variety of congenital cardiac lesions can cause PH (see Table 66.4) (42). The age and type of lesion strongly contribute to the risk of developing irreversible pulmonary vascular disease varies. Early surgery has markedly decreased the development of late PAH and the propensity for postoperative pulmonary hypertensive crises. In general, patients with a VSD or patent ductus arteriosus do not develop irreversible pulmonary vascular changes before 2 years of age. However, without appropriate surgery an estimated 50% of patients with a large nonrestrictive VSD will develop Eisenmenger syndrome (191). Patients with an ASD are less likely to develop severe PAH, and this usually occurs in the third to fifth decade. Infants with an ASD or VSD with concomitant chronic lung disease are at an increased risk for the early development of severe pulmonary vascular disease. In one study of infants with BPD who underwent cardiac surgery for the repair of CHD, 25% of those who died had PAH (192). In the Euro Heart survey of adults with CHD, 882 patients with a secundum ASD were identified. Hemodynamics were worse in nonoperated patients than the patients whose defects had been closed. In patients with unoperated moderate or large defects, clinical parameters tended to worsen with time. In adults with small defects, the patients did well and an operation was not necessarily indicated (193). Eisenmenger syndrome in adults with an ASD was rare and occurred in 15 of 896 patients in a different analysis of the data (194). The risk of PAH with a sinus venosus ASD is likely higher than that of a secundum ASD (195,196). Of the 1,877 patients in the survey, there were 710 with a VSD; the defect was closed in 275, open without Eisenmenger syndrome in 352 and open with Eisenmenger syndrome in 83. Patients with cyanotic congenital cardiac lesions such as transposition of the great arteries, truncus arteriosus, and univentricular heart with high flow are more likely to develop rapid irreversible pulmonary vascular disease. Palliative shunting operations for certain cardiac anomalies designed to increase pulmonary blood flow also may lead to the subsequent development of PH.
Operability
In the child with CHD and pulmonary vascular disease, determination of baseline hemodynamics and reactivity to vasodilators is crucial for successful short- and long-term outcomes (144,146,197,198,199,200,201,202,203,204,205,206). A recent manuscript has extensively reviewed this topic (198). One of the most important factors in long-term survival and freedom from pulmonary vascular disease is the age at which surgery is performed. In 1984, Rabinovitch stated “early corrective surgery is the best safeguard against the persistence or progression of structural changes in the pulmonary vascular bed” (146). In their series, all children repaired before 9 months of age, regardless of severity of the preoperative Heath–Edwards, morphometric, or hemodynamic changes had normal PAP 1 year after surgery. Rabinovitch and Haworth also noted that certain lesions, such as d-transposition of the great arteries, complete AVSD, and truncus arteriosus were more prone to the early development of severe pulmonary vascular changes than others (144,146,203). In general, surgery of the child with CHD is recommended before 2 years of age (207,208,209), but most centers will perform complete repair of the majority of lesions within the first months of life.
Cardiac catheterization aids in determination of pulmonary vascular resistance, pulmonary to systemic vascular resistance ratio, and pulmonary to systemic blood flow. However, several issues arise in the determination of hemodynamics in these patients. Viswanathan and Kumar have described that the assessment of preoperative PVR and pulmonary vascular reactivity in the patient with CHD is difficult and is prone to error (200). Hemodynamic evaluation of these patients is a “snapshot” that may not represent the usual state of the patient. These studies are frequently performed under general anesthesia which frequently leads to a lower systemic blood pressure than exists in the pre-catheterization condition (109). Furthermore, the determination of oxygen consumption in children with CHD is problematic (210,211). Oxygen consumption is frequently estimated as measurement of oxygen consumption in the intubated patients is not readily available due to lack of an FDA-approved device. The LaFarge equation introduces significant overestimation of oxygen consumption in ventilated patients with CHD of all ages, particularly in children younger than 3 years (210,211). Estimation rather than measurement of PaO2 in the pulmonary veins leads to overestimation of pulmonary blood flow, and subsequently underestimation of the PVR. In patients with single ventricle anatomy and physiology additional problems arise. In patients with a bidirectional cavopulmonary anastomosis, the PAP is measured with less than a full cardiac output, thus the pressure after Fontan completion may not be similar. Furthermore, aortopulmonary collaterals are frequent in the single ventricle patient leading to underestimation of pulmonary blood flow by catheterization. Patients with CHD may also have various degrees of pulmonary vein disease leading to inability to accurately measure PVR. This problem, in addition to the low cardiac output seen in pre-transplant
evaluation makes assessment of PVR in the pre-transplant evaluation particularly difficult (212).
evaluation makes assessment of PVR in the pre-transplant evaluation particularly difficult (212).
Although many studies have evaluated hemodynamics to determine operability, the precise values that best correlate with early and late outcome remain unclear. Many of these studies are not directly comparable due to differences in sedation and the use of measured or assumed oxygen consumption. The NHS-1 study of VSD found that no child repaired younger than 2 years of age, regardless of Rp:Rs had an elevated Rp:Rs 4 to 8 years after surgery (209). Haneda et al. reported similar findings (208). In 1982, Lock evaluated the pulmonary vascular response to oxygen in 25 children with large VSDs and elevated PVR. A positive preoperative response to oxygen, defined as a fall in the Rp/Rs of 30%, did not correlate with either operative survival or late Rp/Rs (213). In adult patients with an isolated ASD, Steele retrospectively evaluated patients with a total pulmonary resistance (TPR) greater than 7 U × m2. TPR (defined as mean PAP/pulmonary flow index) had the highest correlation with survival of the hemodynamic parameters measured. Of 18 surgically treated patients at 10-year follow-up, only 1 of 14 with TPR of <15 U × m2, compared with 3 of 4 with TPR of >15 U × m2 had died (195). Bush et al. in 1988 measured PVR and evaluated lung biopsies in 14 children with PHT and CHD. Patients with a lowest pulmonary vascular resistance of greater than 6 U × m2 had a poor prognosis whatever their lung morphology, and some patients with Heath–Edwards grade I or II still had a high PVR. Other vasodilators, such as isoproterenol, were used in a study of 87 children and young adults with VSD. When resting resistance was less than or equal to 7.9 U × m2, it always decreased with isoproterenol and no postoperative problems were experienced in 21 patients (214). In 1991, Moller et al. evaluated 290 patients who underwent closure of a VSD from 1954 to 1960. Death occurred in 59 patients, with higher mortality rates in those operated on after the age of 5 years, those with pulmonary vascular resistance greater than 7 U × m2, and those with complete heart block (Fig. 66.15) (215). More recently, Berner studied 13 children aged 6 months to 16 years with a baseline PVR of >6 U × m2 and Rp/Rs of >0.3 at catheterization with inhaled nitric oxide (iNO). Six of seven with a fall in PVR and Rp:Rs >10% and a value of Rp:Rs <0.3 survived (216). iNO (80 ppm) with oxygen was studied by Atz et al. and showed that a response of 20% or more reduction in PVR was seen in 22/25 patients with NO + O2 compared with 16/25 in O2 alone. Of the 71 patients studied, 10 underwent surgery. In these patients baseline PVRI in room air was 12.9 ± 1.9 U × m2 that decreased to 4.1 ± 1.9 U × m2 in NO (217). In a collaborative study of 10 institutions, 124 patients with CHD were studied under conditions of room air, oxygen, and nitric oxide (NO) (218). Seventy-four of these underwent surgery and 12 died or developed right heart failure. When Rp/Rs of <0.33 was used as the criterion, sensitivity (64% vs. 97%) and accuracy (68% vs. 90%) were increased by using inhaled NO (iNO) in combination with oxygen compared with using oxygen alone (Fig. 66.16). When iNO plus oxygen was used, the lowest value of Rp/Rs that was associated with a poor outcome was 0.16. The highest value of Rp/Rs that was associated with a good outcome was 0.41 (Fig. 66.16) (218). Most recently Kannan et al. in 2003 described long-term follow-up of 38 patients with an unrestrictive VSD and elevated PVR >6 9 U × m2 operated at a median age 7.5 years (range, 6 months to 27 years), from 1985 to 1996 in India. Preoperative pulmonary vascular resistance, ratio of pulmonary blood flow to systemic blood flow, and ratio of pulmonary vascular resistance to systemic vascular resistance were 7.63 ± 1.8 Wood units, 1.9 ± 0.48, and 0.41 ± 0.12, respectively. Thirty patients (79%) had a good outcome and were asymptomatic at a mean follow-up of 8.7 years, with significant reduction in pulmonary artery pressures. Eight patients (21%) had a poor outcome, but there was no significant difference regarding hemodynamic parameters at baseline between those who had a good outcome and those who did not (219). Importantly Kumar et al. recommend a holistic approach that combines clinical, noninvasive, and invasive data to make a decision regarding operability in this challenging group of patients with shunt lesions and elevated pulmonary vascular resistance (200,220). This approach and an algorithm have recently been described by Lopes (Fig. 66.17) (220). Patients who are younger than 2 years of age with evidence of pulmonary
over-circulation and exclusive left-to-right shunting by echo do not necessarily need to undergo cardiac catheterization prior to surgery. Those patients who are older and have desaturation or bidirectional shunting may benefit from vasodilator testing to stratify risk (221). Although there is inadequate long-term follow-up, use of vasoreactivity testing to determine operability suggests that those patients with a PVRI of 6 to 9 Wood units × m2 and PVR:SVR 0.3 to 0.5 may have a favorable outcome if there is a decrease in PVRI and PVR:SVR of at least 20% with a final PVRI of less than 6 Wood units × m2 and PVR:SVR 0.3 (198,206,214,215,222,223,224). Biomarkers, such as circulating endothelial cells (225,226) and micro-RNAs (227,228) have shown promise as additional markers of response to surgical repair.
over-circulation and exclusive left-to-right shunting by echo do not necessarily need to undergo cardiac catheterization prior to surgery. Those patients who are older and have desaturation or bidirectional shunting may benefit from vasodilator testing to stratify risk (221). Although there is inadequate long-term follow-up, use of vasoreactivity testing to determine operability suggests that those patients with a PVRI of 6 to 9 Wood units × m2 and PVR:SVR 0.3 to 0.5 may have a favorable outcome if there is a decrease in PVRI and PVR:SVR of at least 20% with a final PVRI of less than 6 Wood units × m2 and PVR:SVR 0.3 (198,206,214,215,222,223,224). Biomarkers, such as circulating endothelial cells (225,226) and micro-RNAs (227,228) have shown promise as additional markers of response to surgical repair.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


