Pathophysiology of the Heart Failure Clinical Syndrome
Gary S. Francis
Overview
Heart failure has emerged to have a major bearing on public health: In the United States, the numbers are staggering:
995,000 annual hospitalizations for heart failure as a primary diagnosis
2.5 million annual hospitalizations for heart failure as a primary diagnosis
164% increase in hospitalization rate over the past 15 years
12 to 15 million physician visits annually
6.5 million hospital days per year
In-hospital mortality of 5% to 8%
Annual mortality for heart failure of 40% to 60% for some patients
Average patient taking six medications
78% of patients with at least two hospitalizations per year
20% of hospitalized patients rehospitalized within 6 months
Single highest diagnosis-related group in patients more than 65 years of age
Estimated direct costs $23.7 billion in 2004
Two questions come to mind when one is made aware of these statistics: What is “heart failure”? What is the driving force behind this huge increase in incidence and prevalence?
The definition of heart failure is still debated among experts. This lack of clarity stems from differences between historical bedside observations (e.g., tachypnea, cardiomegaly, gallop rhythm, rales, fluid retention) and later laboratory observations regarding muscle mechanics and organ function. It is important to recognize that the historical bedside observations originally used to define heart failure were probably made in severely ill hospitalized patients and as such would represent only a fraction of today’s heart failure population. Textbook signs and symptoms of advanced heart failure do not often pertain to the largest segment of the heart failure population, because most patients today are ambulatory and stable.
Measurement of contractile abnormalities at the organ and molecular levels likewise fails to provide a clear picture of what is wrong with the heart. Part of the problem is the lack of a simple laboratory test that defines heart failure, including plasma β-type natriuretic peptide (BNP). In my view, heart failure should be defined as a clinical syndrome manifested by breathlessness and fatigue at rest or during exertion with accompanying structural and/or functional myocardial disease. In this chapter, I concentrate on patients with major impairment of systolic function. Their hearts are typically remodeled to a more rounded shape, are usually dilated, are often hypertrophied, and by definition are dysfunctional. Mitral regurgitation is frequently evident.
Why are we seeing so much heart failure today when newer therapies that block the renin-angiotensin-aldosterone system (RAAS) and the sympathetic nervous system are claimed to be so effective at reducing mortality? What we are probably seeing is a medically induced delay or forestallment of severe signs and symptoms, but not an actual cure of heart failure. The progression of heart failure may slow down with modern therapy, and the end stages are typically now seen later than before. Heart failure is basically a syndrome that clusters in the elderly.
Unlike thrombolysis for acute myocardial infarction, in which the case fatality rate is reduced early after treatment, in heart failure the Kaplan-Meier survival curves diverge and then later converge. This finding indicates that the benefit is only temporary. Mortality is delayed rather than truly reduced
(1). Unlike in acute myocardial infarction, the natural history of heart failure is one of progressive decline in systolic function. Although the progressive decline may slow in response to specific drug therapy, patients with heart failure usually deteriorate with worsening symptoms, and all eventually die of the disease. Patients are now dying later in the natural history of heart failure. Angiotensin-converting enzyme (ACE) inhibitor therapy prolongs life by only 9 months on average. The addition of β-blockers confers an additional 7-month survival benefit, and spironolactone may provide 12 months of additional survival. However, toward the end of life, many patients express a desire for improved quality of life over increased quantity of life. Their survival may have been prolonged, but some patients have intolerable symptoms and multiple comorbid conditions. From a public health stand point, what we are seeing is older patients with heart failure who are “sicker” with multiple comorbidities and who are in need of multiple therapies.
(1). Unlike in acute myocardial infarction, the natural history of heart failure is one of progressive decline in systolic function. Although the progressive decline may slow in response to specific drug therapy, patients with heart failure usually deteriorate with worsening symptoms, and all eventually die of the disease. Patients are now dying later in the natural history of heart failure. Angiotensin-converting enzyme (ACE) inhibitor therapy prolongs life by only 9 months on average. The addition of β-blockers confers an additional 7-month survival benefit, and spironolactone may provide 12 months of additional survival. However, toward the end of life, many patients express a desire for improved quality of life over increased quantity of life. Their survival may have been prolonged, but some patients have intolerable symptoms and multiple comorbid conditions. From a public health stand point, what we are seeing is older patients with heart failure who are “sicker” with multiple comorbidities and who are in need of multiple therapies.
What Is the Hallmark of Chronic Systolic Heart Failure?
The diagnosis of heart failure remains largely clinical. It is predominantly a bedside diagnosis. However, certain structural features of chronic heart failure are nearly always evident by echocardiography. Systolic heart failure is characterized by progressive left ventricular (LV) dilatative remodeling, and this is perhaps the fundamental lesion or hallmark. The LV chamber increases in size. The heart becomes more spheroidal. Wall tension increases commensurate with LV dilatation. Systolic performance worsens. Stroke volume can be maintained even when ejection fraction is markedly reduced, because end-diastolic volume is increased. It is the LV remodeling that drives the natural history of heart failure under most circumstances, and it is the remodeling that is now the prime target for therapy (2).
Diagnosis and Evaluation of Heart Failure
History and Physical Examination
Breathlessness is the paramount symptom of heart failure (3). It is sensitive but not specific for the disease. It can occur at rest or with minimal physical activity. For most patients with heart failure, feeling breathless is part of everyday life; it is also something for which they develop strategies to prevent or minimize. When the condition becomes “worsening,” it often results in hospitalization (3). The mechanism of dyspnea in heart failure is complex, incompletely understood, and multifactorial (4). It also depends on the context in which it occurs. In acute pulmonary edema, hypoxemia likely contributes to a sense of dyspnea. However, patients with stable chronic heart failure are not usually hypoxemic, but they still are dyspneic. The sensation of dyspnea in the setting of chronic heart failure seems to bear little relation to pulmonary capillary wedge pressure, central hemodynamics, or dead space inhalation (4). Rather, multiple mechanisms cause dyspnea, including respiratory muscle fatigue, increased physiologic dead space, reduced pulmonary compliance, increased airway dysfunction, and perhaps efferent signals from pulmonary J receptors and respiratory muscles (5,6).
Orthopnea, paroxysmal nocturnal dyspnea, and Cheyne-Stokes respirations (7) occur in patients with more advanced heart failure. Cheyne-Stokes breathing carries a poor prognosis (8). Sleep apnea is also common in patients with heart failure and can be associated with an elevated pulmonary capillary wedge pressure (7). Sleep apnea can be central or the result of airway obstruction (9,10). Patients and their spouses should be thoroughly queried about the patient’s sleeping disorder, because it can influence both prognosis and treatment of heart failure. Obstructive sleep apnea can be successfully treated with continuous positive airway pressure in perhaps 60% of cases (11), whereas the treatment of central sleep apnea
may require more aggressive diuretic use in some cases. It is not clear whether continuous positive airway pressure is effective treatment for central sleep apnea. Referral of patients to a sleep laboratory should be considered when sleep apnea or Cheyne-Stokes breathing is suspected.
may require more aggressive diuretic use in some cases. It is not clear whether continuous positive airway pressure is effective treatment for central sleep apnea. Referral of patients to a sleep laboratory should be considered when sleep apnea or Cheyne-Stokes breathing is suspected.
TABLE 85.1 Recommended Tests for Patients with Signs or Symptoms of Heart Failure | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
TABLE 85.2 Echocardiography and Radionuclide Ventriculography Compared in Evaluation of Left Ventricular Performance | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
The second cardinal feature of heart failure is chronic fatigue. Unfortunately, fatigue is also common, very nonspecific, and, like dyspnea, poorly understood. Fatigue in patients with heart failure can result from low cardiac output, but the mechanism of fatigue is likely multifactorial and much more complex. Improving cardiac output does not always improve fatigue (12,13). Skeletal muscle abnormalities (14,15) can lead to general deconditioning. About 15% to 20% of patients with chronic heart failure are anemic. The mechanism of anemia in heart failure is poorly understood, and its treatment is under investigation. Nevertheless, anemia is common, can be associated with fatigue, and is a poor prognostic sign in patients with heart failure.
Tissue congestion occurs in acute and advanced heart failure and can lead to multiple signs and symptoms. These include dyspnea, right upper quadrant pain (acute passive liver congestion), abdominal discomfort from ascites, and heavy legs with difficulty walking as a result of peripheral edema.
The physical findings in heart failure, as classically described in textbooks, are not truly representative of today’s patients. Resting tachycardia, tachypnea, low blood pressure, rales, a gallop rhythm, mitral regurgitation, jugular venous distention, tender hepatomegaly, ascites, and peripheral edema are signs of advanced heart failure. With the modern use of powerful diuretics, ACE inhibitors, and β-blockers, many ambulatory patients have few physical signs. Moreover, the physical findings are rather limited for estimating hemodynamic compromise (16), but they should not be abandoned. The echocardiogram and plasma BNP determination have not replaced the physical examination, but they can facilitate it. Careful examination of the patient with heart failure is particularly important. The physical examination should include inspection of the neck veins with the patient at 45 degrees, a procedure that requires some training and experience. Close inspection of the neck veins on segmental examination is particularly helpful in judging volume status.
Routine Laboratory Tests
The recommendations for laboratory testing in a “new” patient with heart failure have not changed much throughout the years (Table 85.1). Many physicians would add an echocardiogram and a chest radiography, especially for a patient with new-onset heart failure. In fact, the echocardiogram is the cornerstone of evaluation. It provides information regarding chamber size, wall thickness, ventricular performance, valvular lesions, diastolic dysfunction, and regional wall motion abnormalities. The echocardiogram is also used to distinguish systolic heart failure (dilated LV, low ejection fraction) from diastolic heart failure (preserved systolic function). Radionuclide techniques and contrast left ventriculography are still used to assess myocardial function, but echocardiography has become the standard (Table 85.2).
In recent years, plasma BNP and NT-pro-BNP (N-terminal-pro-brain natriuretic peptide) have become available as diagnostic tools for heart failure. Plasma BNP is available as a point-of-care test in most emergency departments, where it is widely used to facilitate the diagnosis of acute decompensated heart failure (ADHF). There is no question that plasma BNP measurement is useful in the evaluation of patients with dyspnea in the emergency department setting (17,18,19). The more important and as yet unanswered question is whether BNP should be measured routinely to guide the diagnosis and management of chronic heart failure (20). Our own data would suggest caution in this regard (21). Physicians should continue to trust their own clinical skills and judgment. Plasma BNP, which is released in response to increased wall stress in both systole and diastole (22), is not a stand-alone test for heart failure. Patients still need to be seen, questioned, and examined. It is true that it may be helpful to monitor BNP or NT-pro-BNP to help understand prognosis and response to therapy, but these strategies have not been critically tested in rigorous clinical trials. However, such studies are now under way.
Factors Known to Precipitate Acute Decompensated Heart Failure
The natural history of heart failure is highly variable and complex (23). What is clear is that patients with chronic heart failure periodically develop ADHF. Invasive ambulatory monitoring suggests that deterioration often begins many days before presentation to the hospital. There seems to be little difference between worsening chronic systolic heart failure
secondary to a precipitant factor and de novo heart failure (24). The Acute Decompensated Heart Failure National Registry (ADHERE registry) has provided some interesting observations regarding hospitalization for ADHF:
secondary to a precipitant factor and de novo heart failure (24). The Acute Decompensated Heart Failure National Registry (ADHERE registry) has provided some interesting observations regarding hospitalization for ADHF:
The median age is 75.2 years.
Twenty-five percent of patients have de novo ADHF, and 75% have a history of chronic heart failure.
More than half these patients have been previously hospitalized for ADHF.
Dyspnea and congestion are the dominant presenting features.
Seventy-two percent of patients have a history of hypertension.
Nearly 50% of patients have a systolic blood pressure higher than 140 mm Hg on presentation.
Twenty percent of patients have atrial fibrillation.
The median length of stay is 4.3 days.
Only 20% are ultimately admitted to a medical or coronary intensive care unit; most are admitted to telemetry units.
Thirty percent of patients have a history of renal insufficiency
Twenty percent of patients have serum creatinine levels greater than 2.0 mg/dL.
Sixty percent of patients have an ejection fraction lower than 40%.
Fifty-eight percent of patients have coronary artery disease.
Forty-four percent of patients have diabetes mellitus.
Only 5% of patients have a pulmonary artery catheter inserted during hospital admission.
The in-hospital mortality is 4%.
Twenty percent of patients are readmitted within 30 days after discharge; 50% are readmitted for ADHF within 6 months following discharge.
Not all patients with ADHF are similar. Two distinct groups seem to emerge. One group has acute pulmonary edema that often responds quickly to therapy, and the other group manifests severe, chronic heart failure before admission that fails to improve quickly. The latter group often develops the cardiorenal syndrome, characterized by a poor response to diuretics and rising blood urea nitrogen and serum creatinine levels.
When patients deteriorate with ADHF, a search should begin for identifiable causes (Table 85.3). A 12-lead electrocardiogram should always be performed to look for myocardial ischemia or infarction. Sequential serum troponin levels are also useful. Superimposed infections, arrhythmias (i.e., rapid atrial fibrillation), and metabolic problems should be sought and aggressively treated. In general, patients with ADHF benefit from hospitalization. According to the ADHERE registry, most patients are admitted to telemetry units and stay about 4 to 5 days. Of some interest, most episodes of ADHF are associated with hypertension and congestion, a finding suggesting that vasodilators and diuretics are the mainstay of therapy, whereas correction of precipitating factors remains an important feature of treatment.
TABLE 85.3 Causes of Acute Decompensation of Chronic Heart Failure | |
|---|---|
|
Mechanisms of Left Ventricular Dysfunction
Abnormalities of Chamber Function
Because virtually any form of heart disease can lead to heart failure, the etiologic basis of heart failure is vast. Coronary artery disease, hypertension, diabetes mellitus, valvular heart disease, and dilated cardiomyopathy are frequently associated with systolic heart failure. The Framingham Study suggests that progression from hypertension to heart failure is still very common (25), and hypertension is frequently present on admission to hospital for ADHF (24). A functional abnormality of systolic heart failure is a diminished ability of the failing muscle to develop force and to shorten at a given velocity and specified loading conditions. Of course, performance may be aggravated by many factors such as noncontractile scar, valvular insufficiency or stenosis, or excessive afterload (i.e., wall stress). There is a decrease in the maximal rate of force development, but generally no major change occurs in the passive length tension or elastic element of heart muscle. However, the passive pressure-volume relation can change dramatically. The failing LV is exquisitely sensitive to afterload conditions (Fig. 85.1).
Several compensatory mechanisms are activated in the heart failure syndrome to adapt myocardial performance to altered loading conditions. Normally, as preload rises and sarcomeres stretch toward their limit of 2.2 μg, contractile force is increased, the so-called Frank-Starling mechanism (Fig. 85.2). The relation between sarcomere length and the development of tension in cardiac muscle is the basis for the Starling law of
the heart. There is a length-dependent activation of cardiac myofibrils by calcium, probably owing to a change in calcium sensitivity of the myofibrils in the extended state. In systolic heart failure, the myocardium fails to generate a normal increase in force development when preload (i.e., sarcomere stretch) is enhanced (Fig. 85.2). Thus, the Frank-Starling mechanism fails to improve myocardial performance to a normal extent in the failing heart (26). In contrast, a diuretic-induced reduction in preload in patients with heart failure does not necessarily reduce stroke volume, just as volume expansion does not raise stroke volume sufficiently. Dogs with experimental heart failure from rapid pacing are unable to mount a normal response to improved preload (27). Alterations in filament-regulatory proteins such as troponin T or troponin I or other isoform changes may play some role (28), or there may be a problem with delivery of calcium to the myofibrils. Despite decades of investigation regarding the cellular and molecular mechanisms of heart failure, there is still no clear understanding of the precise events leading to diminished LV performance (29). Undoubtedly, multiple mechanisms are operative at a cellular level, including regulation of gene expression, imprecisely understood metabolic changes, and important peripheral adaptations. The complexities inherent in the heart failure syndrome and its multiple causes suggest that no single effective therapy will emerge.
the heart. There is a length-dependent activation of cardiac myofibrils by calcium, probably owing to a change in calcium sensitivity of the myofibrils in the extended state. In systolic heart failure, the myocardium fails to generate a normal increase in force development when preload (i.e., sarcomere stretch) is enhanced (Fig. 85.2). Thus, the Frank-Starling mechanism fails to improve myocardial performance to a normal extent in the failing heart (26). In contrast, a diuretic-induced reduction in preload in patients with heart failure does not necessarily reduce stroke volume, just as volume expansion does not raise stroke volume sufficiently. Dogs with experimental heart failure from rapid pacing are unable to mount a normal response to improved preload (27). Alterations in filament-regulatory proteins such as troponin T or troponin I or other isoform changes may play some role (28), or there may be a problem with delivery of calcium to the myofibrils. Despite decades of investigation regarding the cellular and molecular mechanisms of heart failure, there is still no clear understanding of the precise events leading to diminished LV performance (29). Undoubtedly, multiple mechanisms are operative at a cellular level, including regulation of gene expression, imprecisely understood metabolic changes, and important peripheral adaptations. The complexities inherent in the heart failure syndrome and its multiple causes suggest that no single effective therapy will emerge.
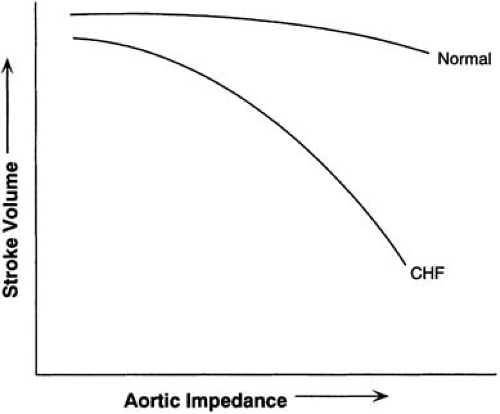 FIGURE 85.1. A hallmark of heart failure is the exquisite sensitivity of the left ventricle to an afterload stress. As impedance to ejection is raised, there is an impressive reduction in left ventricular performance. CHF, congestive heart failure. |
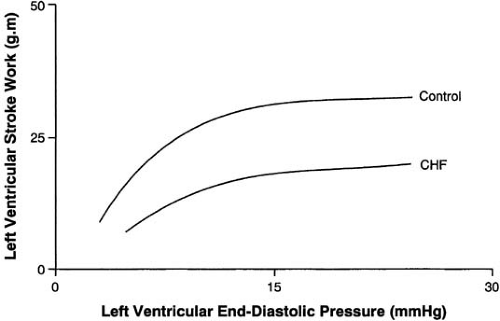 FIGURE 85.2. The Frank-Starling mechanism is altered in heart failure. The failing ventricle is unable to respond to an increase in preload with a normal increase in left ventricular stroke work. CHF, congestive heart failure. |
Left Ventricular Remodeling
The hallmark of chronic systolic heart failure is progressive remodeling of the heart. Remodeling is a change in the size and shape of the heart, usually involving the LV. The increase in LV chamber size during heart failure is by definition is not attributed to acute changes in distending pressure (2). Cardiomegaly eventually occurs and leads to a dilated and thin-walled LV. Although wall thickness may vary, it generally thickens insufficiently to accommodate the increase in chamber dimension, thus further increasing wall tension and reducing LV performance. The mechanisms whereby LV remodeling occurs has been the subject of intensive study (30,31). Multiple mechanisms drive the remodeling process, including myocyte loss, replacement fibrosis, and reactive growth in remaining viable cells (Table 85.4). The early sequence of events at the molecular level are still poorly understood, but there is repeat expression of the so-called “fetal program” leading to elongation of myocytes and an increase in intramyocyte natriuretic peptide synthesis. We have known for years that LV dilation following acute myocardial infarction is an accurate predictor of an unfavorable long-term prognosis (32,33). We now understand that this same process of remodeling occurs progressively in patients with chronic systolic heart failure as a consequence of multiple causes and is linked to a poor prognosis (Fig. 85.3).
The Special Case of Remodeling Following Acute Myocardial Infarction
The classic setting of LV remodeling occurs during and following acute myocardial infarction. Although chronic enlargement of the LV cavity may help to preserve stroke volume in the presence of a very low ejection fraction, it is clear that LV enlargement has very deleterious long-term consequences (31). In acute myocardial infarction, an increase in LV chamber size affords some advantage, at least in the short term.
The extent of LV remodeling in acute myocardial infarction depends on the size of the infarction as well as the location and depth of the injury. So-called “infarct expansion” occurs within hours of a large acute anterior myocardial infarction and is associated with increased mortality (34). Infarct expansion is caused by acute dilatation and thinning of the area of infarction that is not explained by new myocardial necrosis
(35). Wall thinning may result from slippage between myocyte bundles. Infarct expansion is readily detected by echocardiogram (36). The eventual increase in end-systolic volume carries powerful prognostic power, even more than the extent of underlying coronary artery disease. Patency of the infarct-related artery is important in protecting against LV enlargement (37,38). The presence of good antegrade blood flow through the infarct-related artery, by either collateral vessels or reperfusion therapy, protects against abnormal wall motion abnormalities and progressive dilatation of the LV (39,40,41,42).
(35). Wall thinning may result from slippage between myocyte bundles. Infarct expansion is readily detected by echocardiogram (36). The eventual increase in end-systolic volume carries powerful prognostic power, even more than the extent of underlying coronary artery disease. Patency of the infarct-related artery is important in protecting against LV enlargement (37,38). The presence of good antegrade blood flow through the infarct-related artery, by either collateral vessels or reperfusion therapy, protects against abnormal wall motion abnormalities and progressive dilatation of the LV (39,40,41,42).
TABLE 85.4 Factors that contribute to left ventricular remodeling | |
|---|---|
|
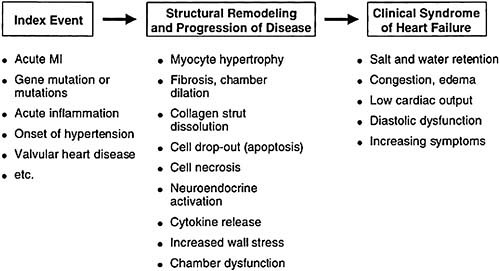 FIGURE 85.3. Progression of left ventricular remodeling in heart failure. MI, myocardial infarction. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


