Chapter 22 Pathophysiology of Renal Artery Disease
Vascular disease affecting the renal arteries presents complex challenges to clinicians. Thanks to recent advances in vascular imaging, more patients than ever before are being identified with some degree of atherosclerotic or fibromuscular renovascular disease. Many of these lesions are of minor hemodynamic importance at the time of detection. Some reach a degree at which perfusion pressures and intrarenal hemodynamics are altered, leading to changes in blood pressure regulation and renal function. These can produce a variety of recognizable clinical syndromes illustrated in Figure 22-1. These range from modest changes in systemic arterial pressure to impaired volume control associated with congestive heart failure (CHF) to threatened viability of the kidney, sometimes designated ischemic nephropathy. Understanding the pathways by which renovascular disease affects cardiovascular and renal disease is important for both diagnosis and for defining optimal management using tools both to block the renin-angiotensin system and to restore the circulation.
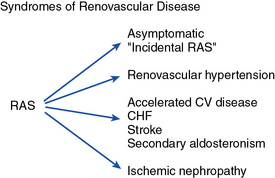
CHF, congestive heart failure; CV, cardiovascular.
(Modified with permission from Garovic V, Textor SC: Renovascular hypertension and ischemic nephropathy. Circulation 112:1362–1374, 2005.)89
Most renovascular lesions are the result of atherosclerosis. With the aging of the U.S. and other Western populations and reduced mortality from stroke and coronary disease, the prevalence of vascular disease in other vascular beds reaching clinically critical levels appears to be increasing.1 Understanding the variety of clinical manifestations of these lesions, the potential for disease progression, and the benefits and limitations of vascular repair are essential for vascular medicine specialists. This chapter will examine the pathophysiology of renovascular lesions regarding blood pressure control, ischemic nephropathy, and clinical syndromes such as flash pulmonary edema. Specific issues regarding diagnostic evaluation and management are addressed elsewhere (see Chapter 23).
A wide range of lesions can affect the renal blood supply, some of which are summarized in Box 22-1. Historically, recognition of renovascular disease resulted from searching for underlying causes of hypertension. This followed the seminal observations of Goldblatt more than 70 years ago2 that renal artery constriction produced a rise in arterial pressure in the dog. These studies were among the first to establish a primary role of the kidney in overall blood pressure regulation. Renovascular hypertension produced by a “clipped” renal artery remains among the most widely studied experimental forms of angiotensin-dependent hypertension.3,4
 Box 22-1 Vascular Lesions That Produce Renal Hypoperfusion and Renovascular Hypertension Syndrome
Box 22-1 Vascular Lesions That Produce Renal Hypoperfusion and Renovascular Hypertension Syndrome
Unilateral Disease (Analogous to Two-Kidney, One-Clip [2K1C] Hypertension)
Epidemiology of Renal Artery Disease
Fibromuscular disease may be identified in 1% to 3% of normal kidney donors subjected to angiography before donor nephrectomy.5 Of those developing clinical hypertension and referred for revascularization, more than 85% are females with a predilection for disease in the right renal artery.6 The location of these lesions is most commonly in the midportion and distal segments of the renal artery. A variety of fibromuscular lesions have been described, but the most common is medial fibroplasia. Occasionally, such lesions may be found in the carotid and other vascular beds, but most commonly they are limited to the renal arteries. Most do not progress to impair renal function, although some lead to arterial dissection and/or thrombosis with loss of the kidney.
Atherosclerosis is the most common cause of renal artery disease. Its presence and severity are related to age and the presence of other atherosclerotic disease of the descending aorta and lower extremities. Population-based series, such as one from North Carolina, indicate that among 834 subjects older than 65 years, significant renal artery stenosis (RAS; defined as Doppler peak systolic velocity (PSV) above 1.8 m/s) can be identified in 6.8% of the general population, regardless of race.7 Recent series of carotid, coronary, and peripheral angiography indicate that the prevalence of renovascular disease corresponds to overall atherosclerotic burden. Incidental renal artery occlusive disease (> 50% stenosis) has been reported in 11% to 18% of patients with coronary artery disease (CAD), particularly when significant hypertension is present.8 Peripheral vascular and aortic disease is associated with higher prevalence (25%-33%). As expected, risk factors predicting the presence of RAS include smoking, hyperlipidemia, hypertension, and diabetes. A corollary observation is that renovascular hypertension resulting from these lesions is now most commonly superimposed gradually upon preexisting essential hypertension. Hence, the blood pressure response and “cure” rates after successful restoration of blood flows to the kidney are limited by preexisting conditions.
Pathophysiological Consequences of Renovascular Disease
Under basal conditions, renal blood flow is among the highest of all organs. This feature reflects the kidney’s filtration function, and less than 10% of delivered oxygen is sufficient to maintain renal metabolic needs. Importantly, a fall in renal blood flow is accompanied by decreased oxygen consumption, partly due to reduced metabolic demands of filtration and tubular solute reabsorption. Reduced renal blood flow can be sustained without measurable change in total kidney oxygen levels (as assessed by renal vein oxygen tension),9 stimulation of erythropoietin release,10 or reduced medullary and cortical tissue oxygenation as measured in human subjects using blood oxygen level–dependent (BOLD) magnetic resonance (MR).11 These observations argue against an overall lack of oxygen as a primary stimulus for either hypertension or renal tissue injury and cast some doubt on the term ischemic nephropathy. Alternative terms proposed included azotemic renovascular disease and hypoperfusion injury.12 Nonetheless, severe vascular stenosis leading to diminished renal perfusion eventually does lead to renal tissue injury and interstitial fibrosis.
Subcritical Levels of Stenosis
The majority of renal artery lesions that compromise renal function are caused by gradually developing atherosclerosis of the renal vascular bed. As noted earlier, some patients undergoing cardiac catheterization have “incidental” renal lesions producing more than 50% cross-sectional stenosis,13 for whom the presence of RAS is a strong independent predictor of mortality. Moreover, nonobstructive RAS (20%-50% decrease in renal arterial luminal diameter) can be found in an additional 28% of patients undergoing cardiac catheterization13 and 48% of patients undergoing aortography for peripheral vascular disease.14 A recent systematic analysis of these reports including more than 15,000 subjects confirmed this overall range of disease prevalence and the rise with increasing atherosclerotic burden.8 Although lesions producing less than 50% in arterial luminal diameter are not considered hemodynamically significant, the relationships between resting pressure gradients and angiographic degree of stenosis are curvilinear and only approximate at best. As a predictor of mortality, even low-grade atherosclerotic lesions denote a hazard nearly equal to more advanced disease.15 Estimating severity of vascular occlusion from angiographic images is notoriously unreliable. It should be emphasized that activation of pressor mechanisms depends upon the presence of a pressure gradient between the aorta and distal renal vasculature16 (Fig. 22-2). There is a general relationship between estimated diameter stenosis and peak translesional pressure gradients, but the relationship is not linear. In some instances, an abrupt fall in poststenotic pressure develops beyond a subcritical range of stenosis.17 Even moderate stenosis, especially when superimposed on intrarenal microvascular disease, may contribute to adverse renal outcomes. Kidneys with a baseline renal artery disease classification of less than 60% stenosis have 11.7% 2-year cumulative incidence of renal atrophy (defined radiologically as a loss of kidney size)18 and 28% cumulative incidence of renal artery disease progression, although progression to total renal artery occlusion is uncommon.19 Increased severity of RAS in patients undergoing cardiac catheterization has an adverse effect on survival, with the 4-year adjusted survival for patients with a 50% stenosis decreasing to 70%, compared to 89% in patients without RAS.20 Therefore, even relatively minor stenosis in the renal artery might have long-term functional implications, especially in the presence of additional risk factors or coexisting renal disease.
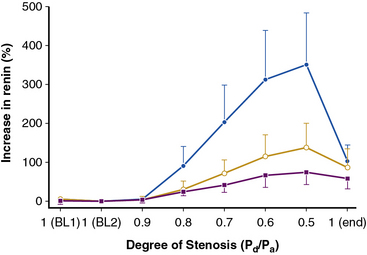
(Reproduced with permission from De Bruyne B, Manoharan G, Pijls NH, et al: Assessment of renal artery stenosis severity by pressure gradient measurements. J Am Coll Cardiol 48:1851–1855, 2006.)16
Renal Microvascular Disease
Lesions in the main renal artery may be superimposed upon or confused with other causes for ischemic renal injury. Intrarenal vascular lesions are commonly observed in the course of various nephropathies, many of which have an ischemic component.21 Risk factors including diabetes, hypertension, atherosclerosis, and aging elicit vasoconstriction or structural changes leading to intrarenal small-vessel disease and ischemic injury similar to that observed in large-vessel disease. Loss of microvessels and impaired capillary repair correlate with development of glomerular and tubulointerstitial scarring,22,23 and may lead to end-stage renal failure. Renal microvascular disease distal to a stenosis in the renal artery may perpetuate and exacerbate renal parenchymal injury and may blunt renal recovery. The presence of small microvessel injury is difficult to verify but may account for changes in diastolic blood flow such as that producing changes in renal resistance index. Elevations of renal resistance index have been proposed to predict poor outcomes in many renal diseases, including renovascular disease.24
Critical Renal Artery Stenosis
High-grade vascular stenosis eventually leads to a decrease in renal perfusion pressure. Critical stenosis is identified when it produces a fall in renal blood flow and glomerular filtration rate (GFR). During experimental renal artery occlusion, the kidney sustains autoregulation of blood flow through a range of perfusion pressures from 200 mmHg to approximately 80 mmHg. Mechanisms underlying autoregulation include myogenic responses to changes in wall tension, release of vasoactive substances, and the tubuloglomerular feedback. The latter responds to decreased renal perfusion pressure and salt delivery by decreasing vascular resistance distal to the obstruction. In addition, during a fall in renal perfusion pressure, the kidney activates multiple pathways that elevate systemic blood pressure, an effect that tends to restore renal perfusion pressure and sustain renal blood flow at the expense of arterial hypertension (Fig. 22-3). Consequently, as long as systemic arterial pressure is allowed to rise, a fall in renal blood flow does not occur until renal arterial diameter is reduced by 65% to 75%. Recent clinical studies suggest that noninvasive radiological imaging commonly overstates the degree of stenosis. Measurement of physiological stimuli, such as the release of renin, indicate that a translesion gradient of at least 10% to 20% reduction is necessary for biological responses to occur in humans.16 To achieve such a gradient, luminal occlusion may need to exceed 80% stenosis. Under some conditions, gradients above 20 mmHg that develop during intrarenal hyperemic challenge with dopamine may disclose hemodynamic significance of lesions under 60% in severity.25
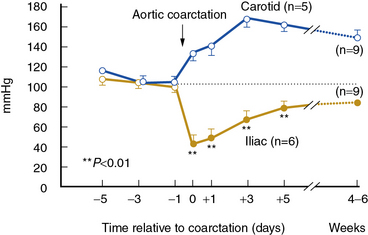
(Reproduced with permission from Textor SC, Smith-Powell L: Post-stenotic arterial pressure, renal haemodynamics and sodium excretion during graded pressure reduction in conscious rats with one- and two-kidney coarctation hypertension. J Hypertens 6:311–319, 1988.)90
When renal perfusion pressure falls gradually, additional mechanisms are recruited that protect the kidney from the functional and morphological consequences observed after acute ischemic injury. These include development of collateral vessels and redistribution of intrarenal blood flow from the cortex to the medulla. Renal cortical blood flow autoregulates more efficiently than the outer medulla, which is continuously on the verge of anoxia. During chronic reduction of renal blood flow, medullary perfusion and oxygenation are relatively maintained by adaptive mechanisms at the expense of cortical blood flow.26 When poststenotic renal artery pressures eventually fall further, either due to progressive vascular occlusion or reduction of systemic blood pressures by drug therapy, renal volume decreases.
In clinical terms, renal atrophy can be defined as a loss of renal length by at least 1 centimeter, and a difference in size between the two kidneys is suggestive of unilateral RAS (or a higher grade of stenosis in one of the kidneys). A decrease in renal volume results from a decrease in filling pressure, filtrate, and blood content of the kidney, as well as structural atrophy of the renal tubules due to apoptosis and necrosis. Apoptosis is an active, pre-programmed form of cell death that is intricately regulated and distinct from cellular necrosis and likely serves as a protective mechanism to allow renal “hibernation.” These changes may be reversible, since tubular cells show vigorous potential for regeneration. Loss of intrarenal microvessels that accompanies the ongoing scarring process may also contribute to renal shrinkage27 (Fig. 22-4), but might be partly reversible upon enhancement of angiogenic signaling.28 However, if a blood flow deficit persists, permanent damage to the kidney may occur. As mentioned, decreased renal blood flow is often accompanied by a decline in GFR and inhibition of tubular epithelial transport that limit renal oxygen consumption and maintain oxygen saturation. Hence, the kidney does not actually develop “ischemia” until an extreme decrease in renal blood flow develops.
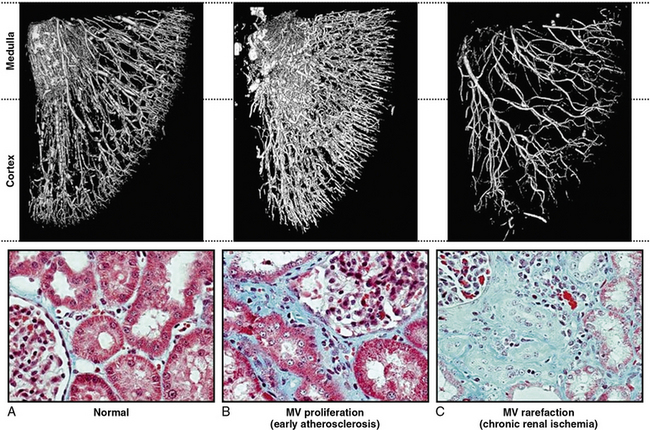
Figure 22-4 Microcomputed tomography imaging of vascular structures in kidney cortex and medulla in a swine model.
(Reproduced with permission from Lerman LO, Chade AR: Angiogenesis in the kidney: a new therapeutic target? Curr Opin Nephrol Hypertens 18:160–165, 2009.)91
Renovascular Hypertension
Goldblatt et al and Loesch were the first to show in the 1930s that obstruction of the renal artery is followed by an increase in systemic blood pressure.2,29 The characteristics of renovascular hypertension depend to a large extent on the status of the kidneys. Unilateral RAS may be present with an intact contralateral renal artery (the experimental form is termed two-kidney, one-clip, or 2K1C). This model is characterized by counterregulatory processes in the contralateral kidney leading to sodium excretion in response to elevated arterial pressure (pressure natriuresis; Fig. 22-5A-B). Alternatively, RAS may affect a solitary kidney (one-kidney, one-clip, or 1K1C; Fig. 22-6A-B). Bilateral RAS and 1K1C lead to more severe renovascular hypertension, although bilateral RAS may behave similarly to 2K1C if one kidney is significantly less ischemic than the other. Patients with this constellation of findings have higher mortality, are more prone to circulatory congestion, and are more likely to experience deterioration of kidney function during administration of antihypertensive agents, including angiotensin-converting enzyme (ACE) inhibitors or angiotensin II (AngII) receptor blockers (ARBs).
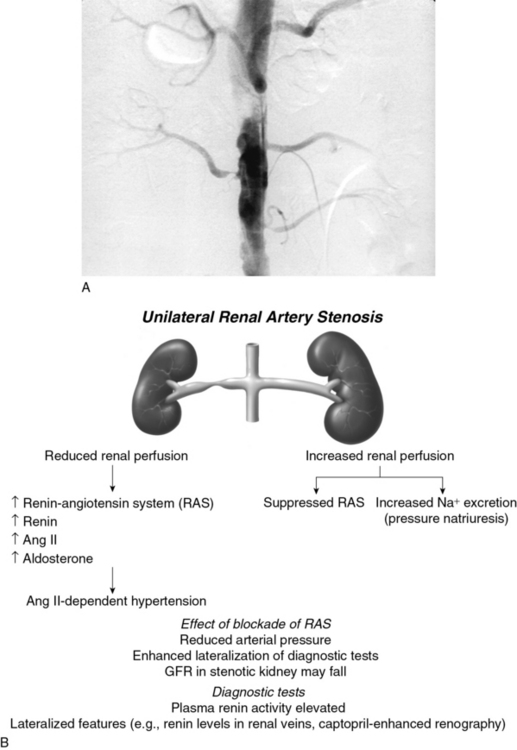
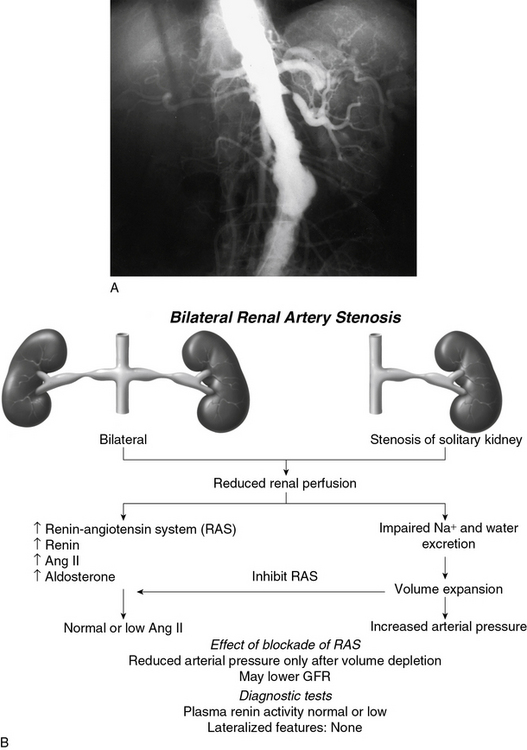
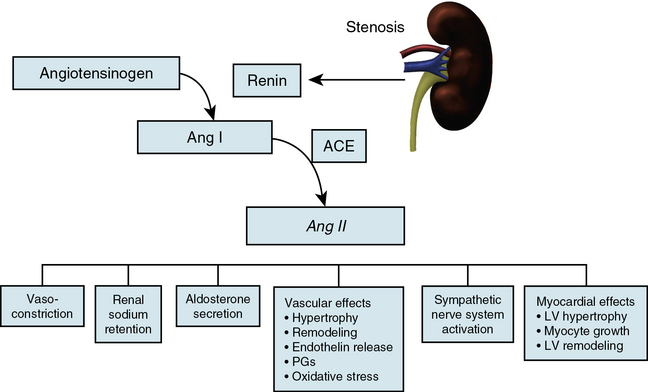
The exact mechanisms responsible for renovascular hypertension have long been debated. The immediate increase in blood pressure in RAS results from release of renin from the stenotic kidney. This leads to increased formation of Ang II, which increases peripheral vascular resistance, plasma aldosterone, sodium retention, extracellular volume, and cardiac output (Fig. 22-7). Early studies using ACE inhibitors30 and more recent studies in an AT1A receptor knockout mouse model of 2K1C confirm the essential role of Ang II in mediating Goldblatt hypertension during its initial phase.31 Experiments with kidney transplantation in these knockout strains indicate that both renal and extrarenal angiotensin receptors participate in regulation of blood pressure.4 Blockade of angiotensin action in experimental models prevents the initial series of events and delays the development of renovascular hypertension indefinitely. Activation of the sympathetic nervous system also plays an important role in the pathogenesis of renovascular hypertension32 primarily via the renal afferent nerves. Both the peripheral and central aspects of the autonomic system are also under the influence of Ang II. If the increase in pressure restores renal perfusion pressure distal to the stenosis, most of these alterations return to baseline levels, with the exception of peripheral vascular resistance.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


